

为促进新药研发成果转化和生产技术合理流动,鼓励药品上市许可持有人、药品生产企业兼并重组,规范药品生产场地变更的注册行为,根据《中华人民共和国药品管理法》《中华人民共和国药品管理法实施条例》等法律法规,食品药品监管总局起草了《药品生产场地变更注册审批管理规定(征求意见稿)》,现向社会公开征求意见。社会各界可于2018年4月19日前,通过登录中国政府法制信息网(网址:http://www.chinalaw.gov.cn),进入首页主菜单的“立法意见征集”栏目提出意见。
药品生产场地变更注册审批管理规定(征求意见稿)
第一条 (法规依据)为促进新药研发成果转化和生产技术合理流动,鼓励药品上市许可持有人、药品生产企业兼并重组,规范药品生产场地变更的注册行为,根据《中华人民共和国药品管理法》《中华人民共和国药品管理法实施条例》,制定本规定。
第二条 (适用范围)已上市中药、化学药品、生物制品制剂的药品生产场地的变更,其注册申请的审评审批适用本规定。
已上市原料药的药品生产场地变更按照相关规定在原料药、药用辅料和药包材登记平台办理相关信息变更登记。
第三条 (变更定义)药品生产场地变更,是指药品的实际生产厂房(包括制造、包装、检验、放行)和生产线等发生改变,包括生产地址的改变或者同一生产地址内生产设施的改变。
同一生产地址,是指负责实际生产的新旧厂房拥有同一物理地址。不同生产地址,是指负责实际生产的新旧厂房拥有不同的物理地址。上述生产地址均应当在药品上市许可持有人或者药品生产企业的批准证明文件中标明。
药品生产地址未变化,但厂房内药品生产条件、生产设备等药品实际生产线发生整体改变的,属于药品生产场地变更。
第四条 (变更原则)药品生产场地变更,一般情况下不应当改变药品上市许可持有人、处方、工艺、药品注册标准;确需改变的,应当按照规定的程序提出补充申请。
第五条 (变更分类)基于药品生产地址的药品生产质量管理规范(GMP)检查历史(包括生产现场检查)、生产地址内所进行的操作以及药品的类别(包括特殊制剂、中药、生物制品等),根据对最终产品可能产生的影响程度,药品生产场地变更分为重大变更、中度变更、微小变更。
重大变更,是指对药品的安全性、有效性或者质量可控性有可能产生潜在较大影响的变更。
中度变更,是指对药品的安全性、有效性或者质量可控性有可能产生潜在影响,但影响较小的变更。
微小变更,是指对药品的安全性、有效性或者质量可控性一般不会产生影响的变更。
第六条 (责任主体)药品上市许可持有人、药品生产企业为药品生产场地变更的责任主体,应当对生产场地变更的必要性及风险进行科学、合理的评估,并按照相关技术指导原则的要求,对变更前后药品质量进行全面分析和研究验证。研究验证应当重点关注变更前后药品质量的一致性。
第七条 (具体程序)药品生产场地发生变更的,药品上市许可持有人或者药品生产企业经评估及研究验证后,应当作为申请人及时提交相应补充申请或者进行年度报告。
属于重大变更的,申请人应当向国家食品药品监督管理总局(以下简称总局)提出药品生产场地变更的补充申请,由总局药品审评机构受理、审评;审评符合要求的,批准变更,发给补充申请批件。
属于中度变更的,申请人应当向总局提出药品生产场地变更的补充申请,总局药品审评机构受理补充申请后在规定期限内未予否定或者质疑的,申请人可以实施该类变更。
属于微小变更的,申请人可以自行实施,同时向总局药品审评机构年度报告相关变更情况。
第八条 (现场检查)申请人提出药品生产场地变更的补充申请后,总局药品审评机构应当结合品种类别、工艺特点以及新生产地址接受GMP检查的历史等因素进行技术审评。
审评中认为需要组织现场检查的,通知总局核查机构开展现场检查并明确具体检查要求。核查机构组织完成现场检查后,应当及时将核查结果反馈药品审评机构。需要药品检验的,药品审评机构通知核查机构进行抽样,指定药品检验机构进行检验并明确具体检验要求。核查机构完成抽样后应当及时将样品提供给指定的药品检验机构,药品检验机构完成检验应当及时将检验结果反馈药品审评机构。
第九条 (简化情形)下列情形的药品生产地址改变(生物制品除外)实行简化注册,申请人可按照中度变更程序进行申报:
(一)变更前后的药品生产地址均属于同一集团,经评估变更前后药品质量保证体系基本一致,生产设备、标准操作规程、人员的生产操作经验等均保持一致,变更后的药品生产地址曾接受并通过总局核查机构组织的GMP检查或者生产现场检查。
(二)变更前后的药品生产地址均属于同一药品上市许可持有人的药品合同生产场地,经评估变更前后药品质量保证体系基本一致,且变更后的药品生产地址曾接受并通过总局核查机构组织的GMP检查或者生产现场检查。
(三)同一药品生产企业异地整体搬迁,经评估搬迁前后药品质量保证体系基本一致。
第十条 (工作时限)药品生产场地变更的补充申请,其审查受理、审评审批工作时限按照《药品注册管理办法》补充申请规定的时限执行。
第十一条 (新药证书)药品生产场地变更的品种具有新药证书且新药监测期未满的,申请人应当提供《新药证书》所有署名单位同意药品生产场地变更的证明文件。
第十二条 (进口药品)进口药品生产地址变更至境内的,境外药品制药厂商应当向总局提出药品生产场地变更的补充申请。对于已获得境内分包装批准的进口药品,其境外药品制药厂商及境内分包装企业应当在相关补充申请获得批准后2年内,提交注销原大包装及分包装批准证明文件的申请。
第十三条 (特殊药品)麻醉药品、第一类精神药品和药品类易制毒化学品不得以技术转让为目的进行药品生产场地变更。
第二类精神药品制剂发生以技术转让为目的的药品生产地址变更的,变更后的药品生产地址应当取得相应品种的定点生产资格,变更前生产地址的该品种定点生产资格应当同时予以注销。
放射性药品发生以技术转让为目的的药品生产地址变更的,变更后的药品生产地址应当取得相应品种的《放射性药品生产许可证》。
第十四条 (实施要求)本规定自发布之日起施行。原涉及药品生产场地变更的有关规定与本规定不一致的,按本规定执行。
附起草说明
为贯彻药品医疗器械审评审批制度改革,落实“放管服”要求,推进药品上市许可持有人制度的试点,促进新药研发成果转化和生产技术合理流动,鼓励药品生产企业兼并重组,提高竞争力,激发市场活力,国家食品药品监督管理总局组织起草了《药品生产场地变更注册审批管理规定(征求意见稿)》(以下简称《规定》)。现将有关情况说明如下:
一、起草背景
药品生产场地变更是药品上市后变更的常见情形之一。已上市药品的技术转让、委托生产、企业兼并重组、异地搬迁、改建扩建等情形均涉及药品生产场地变更,也是药品生产场地变更发生的原因。长期以来,我国一直未对药品生产场地变更的注册管理提出统一的要求,相关要求散见于药品技术转让等有关的文件以及《药品注册管理办法》之中。在具体操作层面上,申请条件和要求、审批程序等不尽相同。
为进一步规范药品生产场地变更注册申请的申报、审评、审批,合理简化注册审批程序,指导并规范申请人开展已上市药品的生产场地变更研究,在现有规定和指导原则的基础上,结合近几年药品生产场地变更的实际情况以及存在的突出问题,制订《规定》。
二、起草过程
《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)发布后,为落实简化药品生产企业之间的药品技术转让程序的要求,总局进行了认真的研究,在借鉴国际先进管理经验的基础上,提出了以药品生产场地变更为核心的一揽子解决问题的思路。成立了起草工作小组,组织药品审评机构、食品药品审核查验中心及部分省局等单位的专家和相关人员召开讨论会,对药品生产场地变更的实际情况及遇到的问题进行讨论,就起草工作进行研讨,于2017年8月形成《规定》草稿。11月,向社会公开征求意见,根据反馈意见进行修改完善。
三、重点问题说明
(一)基于风险的管理思路
药品生产场地变更对药品质量、安全性、有效性可能带来不同程度的影响,从而可能带来一定的风险。药品生产场地的GMP检查历史(接受或者未接受GMP检查)、生产场地内所进行的操作、药品的类别(例如原料药中间体、原料药、特殊制剂、中药、生物制品等)等是决定风险程度高低的主要因素。根据药品生产场地变更对药品可能产生的影响程度,即风险程度的高低,生产场地变更分为重大变更、中度变更和微小变更,食品药品监管部门据此制定相应的管理策略,申请人则据此开展相应的研究。
(二)强化申请人主体责任
由于药品申请人对药品的研发、生产以及产品的性质以及药品生产场地变更对药品质量、安全性和有效性的影响有着最清楚的了解。因此,申请人是变更研究和研究结果自我评估的主体,应当自觉对生产场地变更前后的药品质量、稳定性、生物学等方面进行全面的研究验证。
(三)关于程序简化
《规定》设计的简化程序主要体现在以下几个方面:一是不再要求药品技术转让的转出省食品药品监管部门出具审核意见。二是不再区分新药技术转让与生产技术转让。三是合理简化集团内转移品种等情形的审批程序。对同一集团内药品(除生物制品外)生产场地变更的,如生产设备、标准操作规程、人员具有的生产操作经验等均保持不变,变更后的药品生产场地符合GMP要求,药品上市许可持有人或者药品生产企业在向国家食品药品监督管理总局药品审评机构申报补充申请后,药品审评机构在规定期限内未予否定或者质疑的,即可实施该类变更。这条规定大大缩短了药品上市的周期,同时也倒逼药品上市许可持有人或者药品生产企业重视对生产场地变更开展规范研究。
(四)政策衔接
本《规定》适用于申请人自身拥有的生产场地变更,药品上市许可持有人制度实施过程中的合同生产场地变更,药品技术转让引起的生产场地变更,同时也适用于进口药品的生产场地变更。在印发《规定》的同时,国家食品药品监督管理总局还将同时印发与《规定》配套的技术指导原则。
药品生产场地变更前后药品处方、生产工艺、生产规模等应当保持一致,不应当发生原料药来源、辅料种类、用量和比例,以及生产工艺、工艺参数等影响药品质量的变化。如有提高药品质量,并有利于控制安全性风险的关联变更,应当按照相关的规定和技术指导原则进行研究,并按《药品注册管理办法》有关规定报补充申请。
来源:中国政府法制信息网

为你推荐
 资讯
资讯 广州试点创新药械“医保+商保”同步结算
本次试点依托国家医保信息平台,在22家试点医院实现医保+商保一站式同步结算,通过提供“商业保险创新药械结算清单”,商保理赔金额将一目了然,市民只需支付医保和商保报销后的...
2025-03-28 18:41
 资讯
资讯 揽入首付款2亿美元,恒瑞医药就一款II期临床药物与默沙东达成新合作
近日,恒瑞医药发布公告称,公司与默沙东达成协议,将恒瑞医药的脂蛋白(a)[Lp(a)]口服小分子项目(包括名为HRS-5346的先导化合物)有偿许可给默沙东,默沙东将获得HRS-5346在大...
2025-03-28 16:24

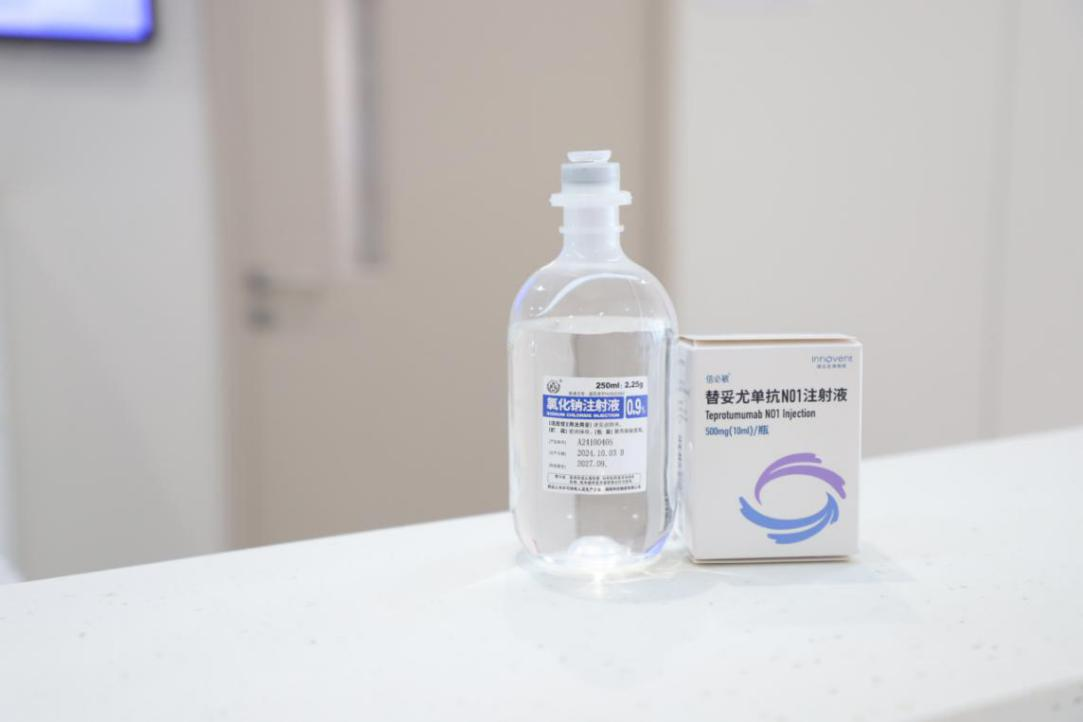 资讯
资讯 国产首款甲状腺眼病靶向药落地湖南,爱尔眼科率先应用
3月27日,爱尔眼科长沙医学中心开出湖南省医院首张国产替妥尤单抗N01注射液处方,并成功为一位中重度甲状腺眼病(TED)患者完成首次注射治疗。
2025-03-27 18:38
 资讯
资讯 复星医药的业绩与生物类似药集采
根据复星医药年报显示,复星医药旗下生物类似药包括第一个国产生物类似药汉利康 (利妥昔单抗注射液)、国内首个获批上市的曲妥珠单抗生物类似药汉曲优 、中国首个中欧双GMP认...
2025-03-27 18:21
 资讯
资讯 预购协议被单方面终止,三叶草生物被要求退还2.24亿美元预付款
3月24日,三叶草生物发布公告,称其全资附属子公司三叶草生物制药(香港)有限公司(以下简称“三叶草香港”)收到全球疫苗免疫联盟(Global Alliance for Vaccines and Im...
2025-03-27 12:10
 资讯
资讯 在华大动作的背后,阿斯利康如何落子“肺健康”
阿斯利康宣布了一项25亿美元的投资计划,在北京建立第六个全球战略研发中心,聚焦于肿瘤、心血管疾病、呼吸系统疾病、免疫学以及人工智能应用等前沿领域的研究和开发,并达成多...
2025-03-27 11:07
 资讯
资讯 罗氏制药与默克达成战略合作,进一步拓展中国肺癌治疗版图
2025年3月26日,罗氏制药和默克共同宣布双方正式签订协议,就特泊替尼(拓得康®)在中国大陆市场的商业化达成合作。双方将充分整合各自优势资源,推动特泊替尼惠及更多METex 1...
2025-03-26 17:17
 资讯
资讯 APASL重磅数据抢先看!吉利德科学公布HBV、HCV、PBC领域多项研究成果
吉利德科学将以壁报和口头报告的形式公布31项肝病领域的最新研究成果,包括慢性乙型肝炎(CHB)领域富马酸丙酚替诺福韦(TAF)的3期临床研究中国队列随访8年的有效性和安全性数...
2025-03-26 14:19
 资讯
资讯 营收飙涨461%现金储备16亿,云顶新耀2024年成功转型Biopharma
3月26日,港股创新药企云顶新耀(1952 HK)发布2024年度业绩报告。报告显示,公司全年收入达7 067亿元人民币,同比增长461%,超额完成了7亿元既定目标。
2025-03-26 10:31


 资讯
资讯 2025“数据要素×”•行业样板发布:华山医院•联仁健康“医院数据资产化生态平台”成功入选
近日,由中国信息协会大数据分会主办,信息化观察网承办的2025数据要素融合与应用创新峰会在北京举办,并正式对外发布了2025“数据要素×”·行业样板100例。
2025-03-25 16:06
 资讯
资讯 全国肿瘤诊疗规范化提升行动启动会在京召开,夯实高质量肿瘤诊疗体系
2025年3月24日,由国家癌症中心学术支持,北京中康联公益基金会、辉瑞公司主办的全国肿瘤诊疗规范化提升行动启动会在北京召开,全国肺癌和肾癌规范化诊疗质控讲师培育计划也于...
2025-03-25 09:52
 资讯
资讯 生态赋能,新加坡助力全球企业迈向国际化新高度
本文将通过三家具有代表性的企业案例,解析新加坡如何利用其完善的产业生态体系,为全球医疗科技企业提供全方位支持,助力其全球化布局。
2025-03-25 09:35


 资讯
资讯 “关注睡眠健康 聚焦抗衰未来”2025睡眠与健康学术研讨会在四川成功举办
在第二十五个“世界睡眠日”到来之时,为促进国民健康、呼吁人们重视睡眠问题、树立“防未病”意识。2025年3月21日上午由中国保健协会健康管理专业委员会与四川天府亨特生命科技...
2025-03-24 16:54

 资讯
资讯 响应“体重管理年”行动,众安互联网医院助力国民科学减肥
在全民健康意识不断提升的当下,体重管理已成为社会各界关注的焦点。随着国家卫健委等部门对体重管理的重视,以及“体重管理年”行动的推进,越来越多的专业机构开始积极探索科...
2025-03-24 11:40