
13日,国家食品药品监督管理总局药品审评中心(CDE)官网发布通知,向社会公开征求《药物注射剂研发技术指导意见》意见。征求意见截止日期:2018年4月15日。
《通知》全文如下:
为进一步贯彻中共中央办公厅、国务院办公厅《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字〔2017〕42号)精神,提高药物注射剂研发的立题合理性,促进药物注射剂研发和生产水平的提升,严格药品注册审评审批技术要求,我中心结合国内外相关指导原则和通用技术指南,起草了《药物注射剂研发技术指导意见》,现向社会公开征求意见。征求意见截止日期:2018年4月15日。
联系人:赵明
电子邮箱:zhaom@cde.org.cn
附:
药物注射剂研发技术指导意见
(征求意见稿)
第一章 总论
第一节 为落实《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》,严格药物注射剂审评审批有关要求,我中心制定《药物注射剂研发的技术指导意见》(以下简称意见)。
第二节 本意见适用于药品上市许可持有人药物研发,也适用于药品审评中心技术审评。本意见适用于化学药品注射剂(包括多组分生化药注射剂)和生物制品注射剂,不适用于中药注射剂。
第二章 药物注射剂总体考虑
第三节 剂型选择基本原则。注射剂可分为溶液型、注射用无菌粉末、注射用浓溶液以及乳剂、混悬剂、注射用油溶液、注射用微球、胶束、纳米粒、脂质体等特殊类型载药系统的注射剂。药物注射剂剂型的研发,以满足临床治疗需求为前提,并综合药物的理化性质、生物学特性等因素,做出科学、合理的选择。
第四节 严格注射剂研发监管。与口服制剂相比,注射剂直接注入人体,是风险程度高的药物剂型。一般情况下,口服制剂已可满足临床需求的,不建议研发注射剂;肌肉注射能够满足临床需求的,不建议再研发静脉注射剂、鞘内注射剂等。对于特殊类型载药系统注射剂的开发,应充分考虑剂型特点和优势,并通过临床试验验证其临床价值及安全性。
第五节 严格注射剂型间转换监管。不鼓励小容量注射剂、大容量注射剂、注射用无菌粉末和注射用浓溶液之间剂型的相互转换。对于剂型间转换应有充分的依据,着重阐述其提高临床价值、产品质量控制的意义。例如,需要说明包括但不限于有效性安全性的变化、治疗顺应性的变化,能够体现出临床优势或明显降低医疗支出成本等。
第六节 对于多组分生化药物和生物制品注射剂,除考虑上述第三、四、五条要求外,还需充分结合自身特点,体现其生物学特性。
第三章 注射剂研发技术要点
第七节 注射剂应围绕疾病治疗需求并结合药物理化学性质合理研发。对于改规格、改剂型、改盐基注射剂的开发,从药物的安全性、有效性、稳定性考虑,应具有明显的临床优势。
第八节 药学方面须满足的基本要求
药物注射剂研发的药学方面须满足如下基本要求:
一是处方要求。注射剂所采用的原料药、辅料、包装容器/材料须符合供注射用的质量要求,并进行登记、公示,与制剂共同审评。对于原料、辅料、包装容器/材料应建立完善的质量保证体系,在杂质(包括元素杂质等)、微生物限度、热原/细菌内毒素等方面应建立严格的控制措施和标准。药物和辅料、包材的相容性应符合要求。对于仿制药应尽量按照与参比制剂一致的原则选择原料药、辅料,通过质量源于设计的原理进行处方、工艺研究;对特殊类型注射剂,需客观的、有针对性的评价原料、辅料和包装材料间的相容性,并结合工艺过程确认辅料的性质对制剂特性的影响。
注射剂中应对抑菌剂的使用严格控制,原则上不建议使用抑菌剂。必须加入抑菌剂的注射剂,应建立抑菌剂的质量要求,并按照中国药典现行版要求提供抑菌效力验证结果及安全性的综合评价。除另有规定外,静脉注射、鞘内注射、硬膜外注射等注射剂,不得添加抑菌剂。
二是工艺要求。灭菌/无菌生产工艺是注射剂的关键工艺步骤,须依据灭菌决策树选择合理的灭菌/无菌生产工艺,对灭菌/无菌工艺进行验证,并提供在拟定的工业生产规模生产线进行的工艺验证方案和报告。关注特殊类型注射剂生产过程中影响制剂特性的工艺因素,对生产工艺进行验证,并提供在拟定的商业化生产线进行的工艺验证方案和报告。申报上市的注册批应该确实保证产品和工艺过程具有工业生产规模下的可行性,并提供验证资料。对特殊类型注射剂,注册批生产规模一般至少是工业生产规模的五分之一或至少生产1000支/瓶,两者中选较大者。对工业生产规模少于1000支/瓶的特殊品种,注册批生产规模应与工业生产规模一致。
三是药物包材相容性。对于直接接触药品的包材,重点关注因素包括容器的保护作用、相容性(详见化学药品与弹性体密封件相容性研究技术指导原则(20161228)、化学药品注射剂与塑料包装材料相容性研究技术指导原则(试行)、化学药品注射剂与药用玻璃包装容器相容性研究技术指导原则(试行))、安全性与功能性,注射剂所用包材应在满足临床使用需要的同时与药物及辅料具有良好的相容性。对于玻璃容器应关注药液对玻璃的侵蚀。
四是质量研究、质量标准和稳定性。应研究注射剂的原料、辅料、包装材料的质量,建立质量标准。对制剂应进行全面的质量研究和稳定性(趋势)研究,在确保无菌、热原/细菌内毒素符合要求基础上,结合具体制剂品种的特点开展针对性的质量研究并制定相关质量标准。
质量及质量标准应关注工艺过程、使用过程中注射剂的质量(物理、化学和生物学特性)。
注射剂稳定性研究内容包括影响因素试验、加速试验和长期试验,必要时应进行中间条件试验考察。对低温下可能不稳定的注射剂建议进行低温试验和冻融试验。对于临用前要调配的产品,应研究调配过程及其调配物的稳定性,确保调配、使用过程中药物的安全性、有效性。对注射剂或调配物与给药器具的相容性、给药过程中给药器具对药物的吸附进行研究。
五是特殊类型注射剂。应研究药物释放等与安全性、有效性、质量可控性有关的指标并建立相应标准,必要时应通过动物试验放行,并提出替代方法;应考察制剂的形态学等物理特性对制剂安全性、有效性、稳定性的影响。鼓励进行制剂体内释放及处置过程的研究。
第九节 临床使用方面须满足的基本要求。
为便于临床使用和控制风险,必须针对临床使用过程各个环节的特殊要求,开展全面系统的研究,并将其纳入说明书相关项下,指导临床用药。
以静脉滴注的粉针剂为例,需要说明包括但不限于其溶解环节的溶媒选择和溶解方法;稀释环节的稀释液选择(应关注稀释液的pH值、渗透压等)和操作方法、操作环境、药物相互作用及配伍禁忌,稀释后的贮存条件及贮存时间,以及使用前检查要求;给药部位选择、滴注浓度及速度、多次给药时留置针的处置,给药过程中及给药后的注意事项,以及多路静脉通道的相互影响;药物配伍稳定性;患者用药须知等。对于儿童用制剂,尚需有准确的剂量分割方法等。
其他制剂应结合制剂及其工艺的特点进行相关研究。
第四章 其他要求
第十节 严格注射剂说明书管理。说明书中用法用量,应遵循能采用口服途径给药的尽量不采用注射途径给药的基本原则。在注射剂产品说明书中,应标明在临床紧急救治或不能经口服用药情况下使用,或作为暂时不能经口服用药的临时替代等提示性内容。
来源:新浪医药新闻

为你推荐
 资讯
资讯 国家药监局发布医药代表管理办法(征求意见稿)
医药代表应当具有医学、药学或相关专业本科及以上学历(或者中级及以上专业技术职称);具有药品临床理论知识及实践经验,或者具有药品研发、生产、检验、质量管理等岗位工作经...
2024-11-29 18:50
 资讯
资讯 三高共管,三慢共防:2024中国医药城大健康产业论坛“慢病协同共管”专题圆满举办
大会以“三高共管,三慢共防”为主题,聚焦共病共管、心肾保护、跨界融合、慢病管理创新模式、创新产业技术等前沿内容
2024-11-29 15:03


 资讯
资讯 湖南省视觉功能及儿童眼病学术研讨会暨世界近视眼大会成果宣讲会在长沙举办
11月28日,由爱尔眼科湖南省区视光学组、爱尔眼科湖南省区小儿眼科学组主办、长沙爱尔眼科医院承办的“湖南省视觉功能及儿童眼病学术研讨会暨世界近视眼大会成果宣讲会”,在长...
2024-11-29 09:09
 资讯
资讯 一个手功能康复训练仪器的设计
系统由3个模块组成:指示锻炼情况和治疗持续时间,并为气动螺纹管提供动力的驱动泵;穿戴在健康一侧手臂的镜像手环,及穿戴在患肢的康复机器手套。
2024-11-29 09:02
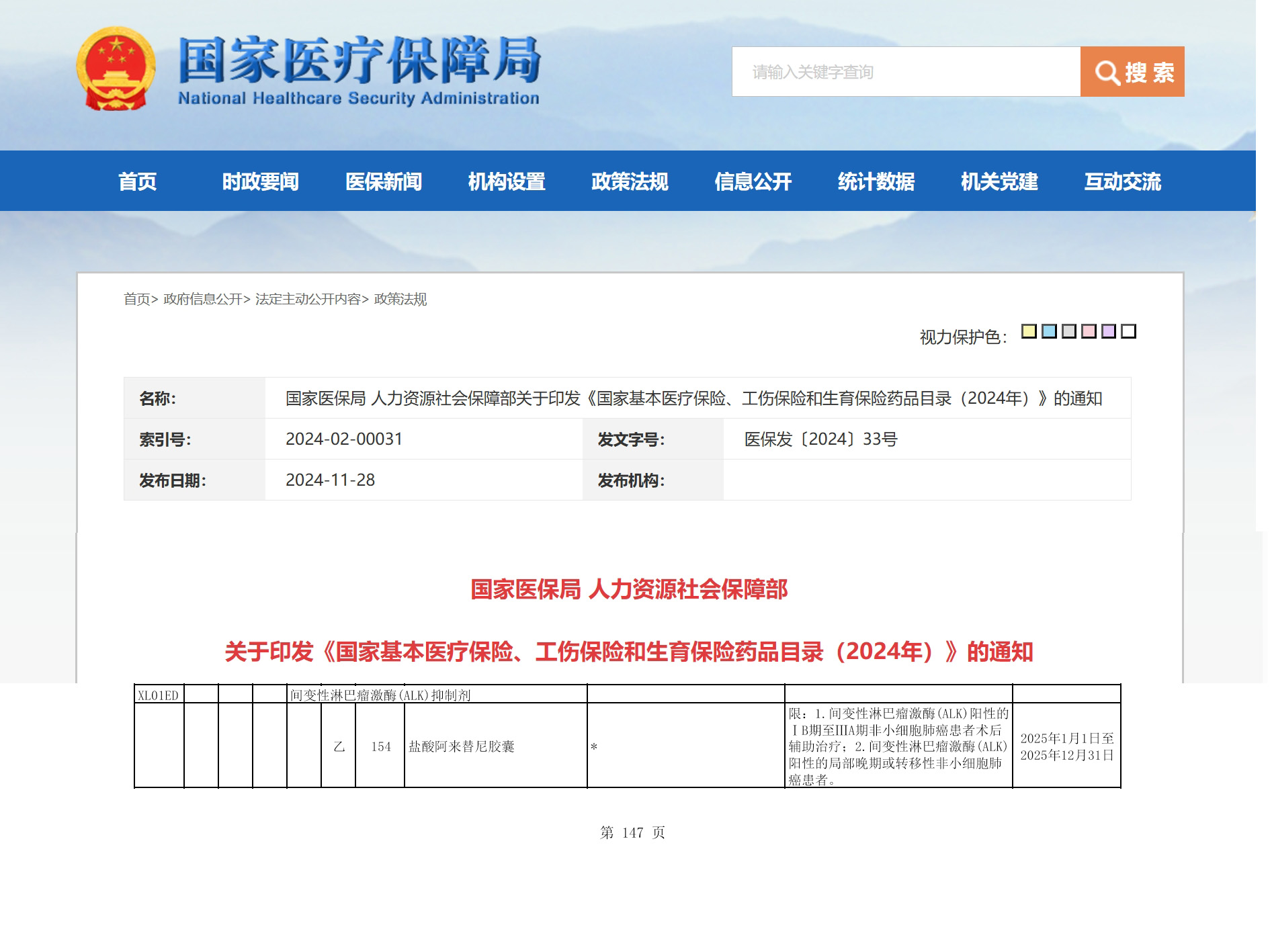 资讯
资讯 阿来替尼ALK阳性早期NSCLC术后辅助治疗适应症成功纳入医保
2024年11月28日,国家医保局公布了《国家基本医疗保险、工伤保险和生育保险药品目录(2025年)》(以下简称:国家医保目录),安圣莎®(通用名:阿来替尼)ALK阳性早期非小细胞...
2024-11-28 18:58

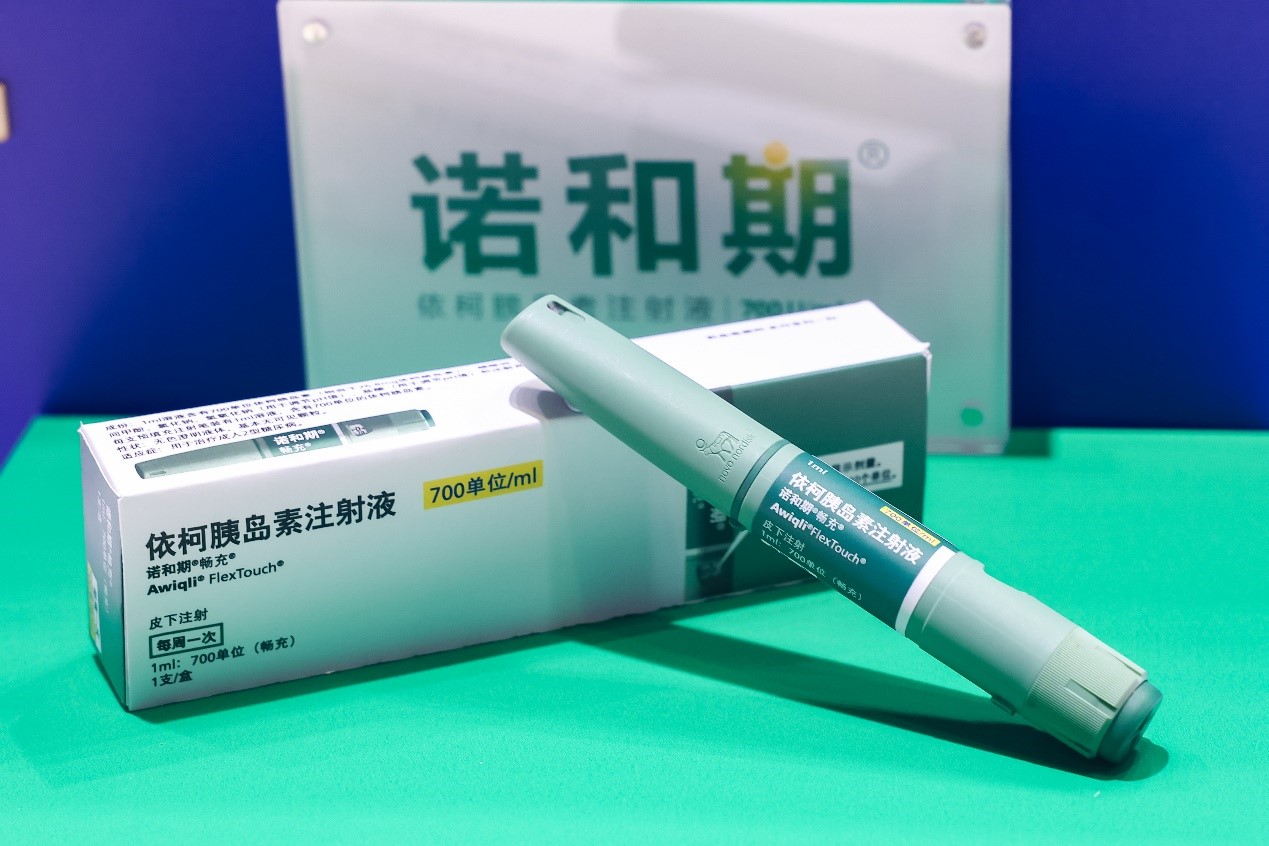 资讯
资讯 全球首个胰岛素周制剂诺和期(依柯胰岛素注射液)成功纳入国家医保目录
11月28日,根据国家医疗保障局最新公布,依柯胰岛素注射液(商品名诺和期®)被纳入《国家基本医疗保险、工伤保险和生育保险药品目录(2024年)》。目录将于2025年1月1日起正式执行。
2024-11-28 15:52
 资讯
资讯 一款国产PD-1被首次被纳入,多款“全球新”在列,2024国家医保谈判结果发布(附完整版目录下载链接)
今日(11月28日),2024年版国家医保药品目录调整名单正式公布。
2024-11-28 14:49
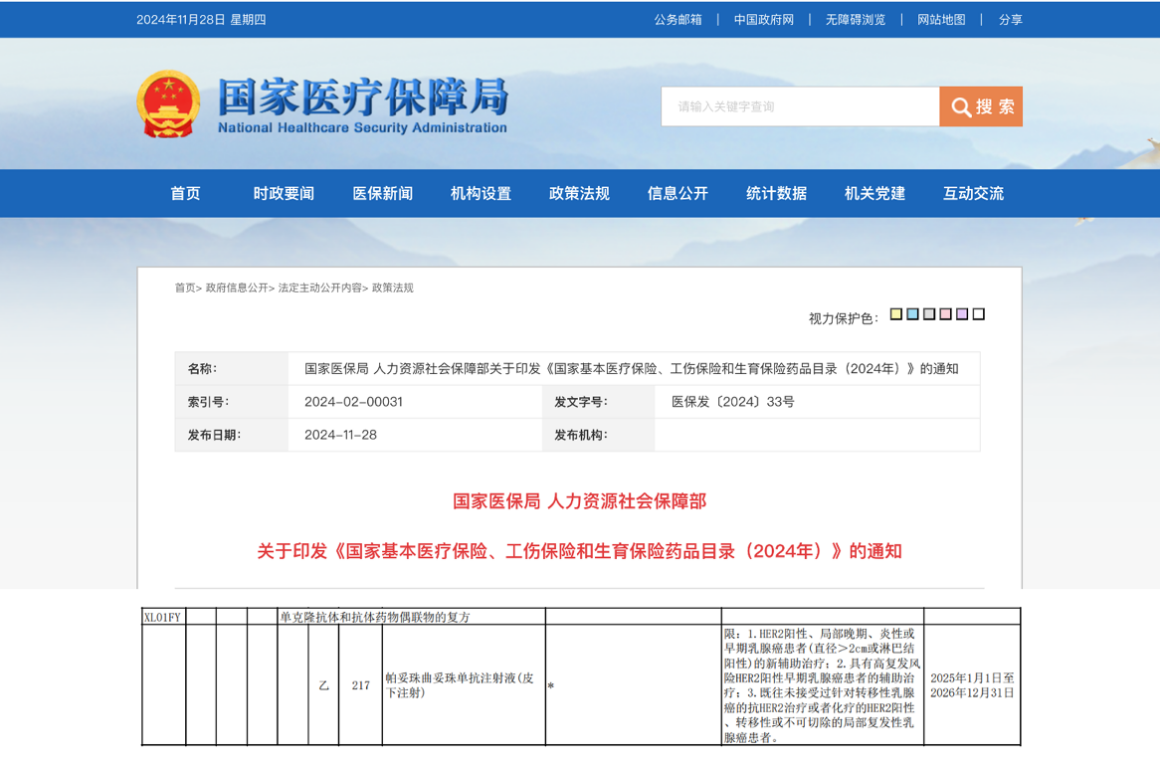 资讯
资讯 首个抗HER2复方皮下制剂赫双妥纳入国家医保目录,5-8分钟靶向治疗助力乳腺癌患者拥抱自由新生
2024年11月28日,国家医疗保障局发布最新公告,罗氏抗HER2乳腺癌皮下生物制剂赫双妥®(通用名:帕妥珠曲妥珠单抗注射液(皮下注射))正式进入《国家基本医疗保险、工伤保险和...
2024-11-28 13:53


 资讯
资讯 罕见病创新药盐酸伊普可泮胶囊纳入2024年国家医保药品目录
作为全球首个且唯一获批的特异性补体旁路B因子抑制剂和成人阵发性睡眠性血红蛋白尿症(PNH)口服单药疗法,纳入医保会大大提升患者用药可及性及便利性,为PNH患者带来新的治疗选择。
2024-11-28 13:09
 资讯
资讯 强生2款创新药物纳入新版国家医保目录,惠及更多中国患者
今日(11月28日),在国家医疗保障局公布的《国家基本医疗保险、工伤保险和生育保险药品目录(2024版)》(以下简称:国家医保目录)中,强生的2款创新药物收获了好消息,包括兆...
2024-11-28 12:25
 资讯
资讯 深化创新产品布局 远大生命科学集团获得幽门螺杆菌创新治疗药物独家商业化权益
根据协议条款,远大生命科学将独家负责TNP-2198在中国大陆、香港、澳门的市场推广和商业化销售。丹诺医药将继续负责TNP-2198针对幽门螺杆菌的后续全部临床、非临床和药学研究、...
2024-11-28 09:28
 资讯
资讯 2024青年防艾公益行动在京启动,防艾主题艺术作品在全国11座城市亮相
在第37个“世界艾滋病日”到来之际,由中国性病艾滋病防治协会与中国青年报社共同主办、吉利德科学公益支持的“2024青年防艾公益行动”正式启动。
2024-11-27 18:51

 资讯
资讯 推动智能实验室进化,安捷伦在Analytica China 2024展示技术与服务创新
近日,第12届慕尼黑上海分析生化展隆重举行,安捷伦科技公司如期亮相。本届展会,安捷伦以“创新不止,深谙所需”为主题,重点展示一系列创新的技术与服务,借以诠释公司对科学...
文/史士云 2024-11-27 17:21
