
昨夜,FDA官网挂出对仿制药巨头梯瓦制药中国萧山工厂的警告信,缺陷包括关键工艺参数未监控、产品质量反复不合格、根本原因分析和CAPA无效、未能建立一个科学合理的取样计划、工艺验证取样不合理、未描述持续工艺确认计划如何确保质量属性持续符合并且批批一致
具体如下:
The U.S. Food and Drug Administration (FDA)inspected your drug manufacturing facility, Teva Pharmaceutical and Chemical(Hangzhou) Co. Ltd., 1889 Jingliu Road, Xiaoshan, Hangzhou, Zhejiang, Chinafrom September 26 to 29, 2016.
FDA于2016年9月26-29日检查了你们位于杭州萧山的梯瓦医药化工有限公司生产基地。
This warning letter summarizes significant deviationsfrom current good manufacturing practice (CGMP) for active pharmaceuticalingredients (API).
本警告信概述了原料药生产严重违反CGMP的行为。
Because your methods, facilities, or controls formanufacturing, processing, packing, or holding do not conform to CGMP, your APIare adulterated within the meaning of section 501(a)(2)(B) of the Federal Food,Drug, and Cosmetic Act (FD&C Act), 21 U.S.C. 351(a)(2)(B).
由于你们原料药的生产、加工、包装或保存的方法、场所或控制不符合CGMP要求,你们的原料药根据FDCA的501(a)(2)(B)以及21 U.S.C.351(a)(2)(B)被认定为掺假。
We reviewed your October 21, 2016 response in detailand acknowledge receipt of your subsequent correspondence.
我们详细审阅了你们公司于2016年10月21日及之后的回复。
During our inspection, our investigator observedspecific deviations including, but not limited to, the following.
我们的检查人员发现的具体问题包括但不仅限于以下:
1. Failure to establishwritten procedures to monitor the progress and control the performance of processing steps that may cause variability in the quality characteristics ofyour API.
未能建立书面程序以监控工艺步骤进程及表现,因此可能会导致你们API的质量特性的波动。
Our inspection found that approximately 10 percent of (b)(4)API batches produced at your facility from December 2014 to September 2016failed to meet the (b)(4) impurity limit. During this period, anadditional 10 percent of batches yielded out-of-trend (OOT) results for (b)(4).You have reprocessed rejected out-of-specification OOS batches but failed toimplement effective corrective and preventive actions (CAPA) to correct processdesign and control flaws that lead to excessive formation of this impurityduring processing.
我们检查发现你工厂自2014年12月至2016年9月生产的约有10%的某API批次不符合某杂质限度要求。在此期间,同一产品另有10%批次质量超趋势(OOT)。你们对OOS批准进行了返工,但未实施有效的CAPA来纠正工艺设计,控制工艺中导致此杂质过量生产的工艺瑕疵。
According to your response, a new root cause analysisfound that impurity failures appear to be related to insufficient control of (b)(4).You committed to monitor (b)(4) specific process parameters in the newprocess performance qualification batches of (b)(4) API and the first (b)(4)commercial batches. However, these proposed parameters differ from the“critical process parameters” monitored by your firm in the last three years.They also do not include all of the parameters that you categorized as“critical and significant” in the most recent process qualification study. Yourresponse does not commit to monitor future batches for all parameters thatimpact quality, and may contribute to the failure of a batch of intermediatesor API to meet specifications.
根据你们的回复,在新的根本原因分析中发现杂质失败貌似与对XX控制不充分有关。你们承诺会在该API新的工艺性能确认批次及首次XX商业批次中监测该特定工艺参数。但是,这些所提出的参数并不是你们公司在过去三年所监测的“关键工艺参数”。他们也不包括所有的你们在最近工艺确认研究中定为“关键和重要”的参数。你们的回复没有承诺在将来监测所有影响质量、可能会与中间体或API不符合质量标准有关的参数。
Your response is also inadequate because it did notinclude the risk assessment and related scientific rationale to ensure thatcontrols implemented for all batches will detect upstream processing variationand ensure final API quality. You also acknowledged in March 2017correspondence that additional lots have failed since you resumed commercialmanufacture of (b)(4) API. Recurrence of product quality failuresfollowing the completion of your investigation and process re-qualificationindicate that your root cause analysis and CAPA were ineffective.
你们的回复不充分还因为其中未包括风险评估以及相关的科学合理性,以确保对所有批次所实施的控制能检出上游工艺波动,确保最终API质量。你们在2017年3月的信函中告知我们自从你们恢复某API的商业生产以来,又有一些批次不合格。在你们完成调查和工艺再确认之后,产品质量又发生不合格表明你们的根本原因分析和CAPA是无效的。
In response to this letter:
在回复此函件时:
Provide an updated investigation into the root cause(s) of (b)(4) OOS results and an improved CAPA plan. Include provisions to ensure CAPA effectiveness.
请提交更新后的某OOS结果根本原因调查,以及改进后的CAPA计划。包括确保CAPA有效性的条款。
Specify if the presumed root causes for failures were actually observed in the failed (b)(4) batches.
说明在不合格的XX批次中是否实际观察到了所假定的根本不合格原因。
Describe why some finished (b)(4) API batches yielded OOS results for the bis-ether impurity, but passed testing for this same impurity at the(b)(4) stage.
说明为什么有些某API批次产生二醚杂质OOS结果,但却能通过在某工艺步骤中该相同杂质的检测。
List the past and current process parameters for (b)(4) API. Explain their role in the process, the potential impact on quality, the limits used, and your justification if you plan to cease monitoring and controlling any parameter during commercial batch manufacture.
列出过去和现在的某API工艺参数。解释在工艺中各参数的作用、对质量的潜在影响、所使用的限度以及如果你们计划停止在商业批次生产中监测和控制任一参数时你们的论证。
Explain your systems for incorporating reprocessing activities into Drug Master Files.
解释你们将返工活动整合入DMF文件的系统。
Provide procedures that ensure that reprocessed lots and process performance qualification lots are included in your stability program.
提供程序确保返工批次和工艺性能确认批准被放入你们的稳定性计划中。
2. Failure to establish a sampling plan based on scientifically-sound sampling practices.
未能建立一个科学合理的取样计划
Our investigator documented deficiencies in your validation sampling plan for (b)(4) API. You did not conduct adequate monitoring and testing during process performance qualification stage to evaluate whether product quality was uniform throughout each batch. You only assessed water content at the drying step for homogeneity.
我们的检查员发现了你们对XX API的验证取样计划的缺陷。你们在工艺性能确认阶段没有进行充分的监测和检验以评价每一批次的产品质量是否一致。你们只在干燥步骤评估了水分含量的均匀性。
In your response, you acknowledged that a higher level of sampling during the revalidation of the manufacturing process revealed some inter-batch variability in residual solvents and particle size distribution of (b)(4).
在你们的回复中,你们确认了在生产工艺再验证期间的一个高水平的取样显示XX的残留溶剂和粒度分布存在一些批内差异。
Your response is inadequate because it did not describe how your continued process verification program assures that quality attributes continue to be met batch-to-batch, as well as uniformly throughout each batch. Regarding uniformity, using only (b)(4) samples for attributes that may significantly vary within a batch is insufficient to ensure that your process remains in an ongoing state of control.
你们的回复是不充分的,因为没有描述你们的持续工艺确认计划如何确保质量属性持续符合并且批批一致。关于一致性,仅仅使用XX样品来确认可能在批内显著变异的属性是不足以确保你们的工艺保持在一个持续受控状态的。
In response to this letter:
回复此函:
· Specify how you will improve batch sampling of (b)(4) API to ensure that you detect intra- and inter-batch variability during commercial manufacturing.
详细说明你们将如何改善XX API的批取样来确保你们可以在商业生产过程中发现批内和批间变异。
· Evaluate other API produced by your firm for adequacy of sampling plans.
· 评价贵司生产的其他API取样计划的充分性。
· Provide overall quality system improvements to ensure all sampling performed by your firm is representative and able to detect non-uniformity of the quality attributes that may vary within a batch.
· 提供质量体系的总体改善以确保贵司执行的所有取样都能具有代表性并能够检测到可能发生变异的质量属性的不一致性。
来源:新浪医药新闻 作者:GMP办公室

为你推荐
 资讯
资讯 恒瑞医药双靶点GLP-1/GIP瑞普泊肽注射液中国T2DM口服药控制不佳人群Ⅲ期研究取得积极顶线结果
近日,恒瑞医药宣布,GLP-1 GIP双重受体激动剂瑞普泊肽(HRS9531,大中华区外称KAI-9531)注射液治疗经二甲双胍单药或联合SGLT2抑制剂治疗后血糖控制不佳成人2型糖尿病患者的中...
2026-08-07 17:23
 资讯
资讯 韩宇药业提交了替尔泊肽的国内首仿上市申请
据国家药审中心官网显示,8月6日,翰宇药业申报的替尔泊肽注射液上市申请获得受理,注册分类为4类(境内外均已上市的药品的仿制药)。从目前公开信息看,这是国内首个替尔泊肽注...
2026-08-07 16:35
 资讯
资讯 礼来半年报业绩大增,二次上调全年业绩指引
8月5日,礼来公布了2026年上半年业绩——总营收427 73亿美元,同比增长49%。其中,第二季度营收229 74亿美元,同比大增48%。此前,礼来第一季度实现营收197 99亿美元,同比增...
2026-08-07 15:57
 资讯
资讯 全球首款 mRNA 季节性流感疫苗诞生!Moderna mFLUSIVA 获 FDA 批准
美国食品药品监督管理局(FDA)正式批准 Moderna 研发的 mRNA 季节性流感疫苗mFLUSIVA(mRNA-1010)上市,用于 50 岁及以上成人预防季节性流感
2026-08-07 13:07
 资讯
资讯 荣昌生物2026年半年度业绩
根据预告,荣昌生物预计 2026 年半年度实现营业收入约 58 5亿元,与上年同期相比,将增加收入约 47 52亿元,同比增加约 433%。预计 2026 年半年度实现归属于母公司所有...
2026-08-06 21:31

 资讯
资讯 信达生物2026上半年产品收入超2024年全年
公告显示:2026年上半年,信达生物产品收入实现超人民币82亿元,同比增长55%以上。其中,公司第二季度产品收入录得超过人民币43亿元,同比增长约60%。根据信达生物此前发布的信...
2026-08-06 15:09
 资讯
资讯 百济神州2026上半年业绩大增,上调全年业绩指引
2026年半年度公司营业总收入222 20亿元,较上年同比上升26 8%;2026年半年度归属于母公司所有者的净利润32 71亿元,较上年同比上升627 1%。
2026-08-06 14:14
 资讯
资讯 因美纳MiSeq i100 Series-CN中国上市,以全球品质与本土制造支持中国客户测序能力建设
MiSeqTM i100 Series-CN正式在中国上市,首批由因美纳中国生产制造基地生产的本土化产品已于近期完成客户交付。
2026-08-06 13:33
 资讯
资讯 石药集团与阿斯利康成立合资公司,收到1000万美元里程碑付款
双方将在河北石家庄建设新一代生物制剂生产基地,该合资公司的初始业务将聚焦于为全球市场生产及供应双方约定的生物制剂药物原液(DS)。
2026-08-05 20:49
 资讯
资讯 全球首款!核心医疗人工心脏产品获欧盟CE认证
Corheart®6 可用于桥接心脏移植、短期心肌功能恢复治疗以及长期终点治疗,完整覆盖终末期心衰从短期生命支持到永久循环辅助的全场景临床需求
2026-08-05 20:37
 资讯
资讯 国家药监局批准3款创新医疗器械上市
近日,国家药品监督管理局批准了河南赛美视生物科技有限公司的“三焦点环曲面人工晶状体”和深圳北芯医疗科技有限公司的“心脏脉冲电场消融仪”、“一次性使用心脏脉冲电场消融...
2026-08-05 16:47
 资讯
资讯 80亿美元,全球两家核药公司合并
美东时间8月3日,Curium Pharma宣布与Lantheus Holdings, Inc 达成最终合并协议。Curium将以每股102 5美元现金收购Lantheus全部流通股,基础对价70亿美元,若2030年前达成...
2026-08-05 13:51
 资讯
资讯 章科:坚持为人民出政绩 推动医疗保障事业高质量发展
医疗保障制度依法保障每一位参保群众的医保权益,有力提高全社会医疗服务的可及性,有助于增进民生福祉、缩小贫富差距,促进社会公平和共同富裕。
2026-08-05 10:48
 资讯
资讯 康宁杰瑞与PathosAI达成22亿美元ADC授权协议,1.25亿美元首付款
8月4日,康宁杰瑞生物制药宣布其全资子公司江苏康宁杰瑞生物制药有限公司与美国临床阶段生物技术公司Pathos AI就TROP2 HER3双特异性抗体偶联药物JSKN016达成独家授权许可协议。
2026-08-04 22:14
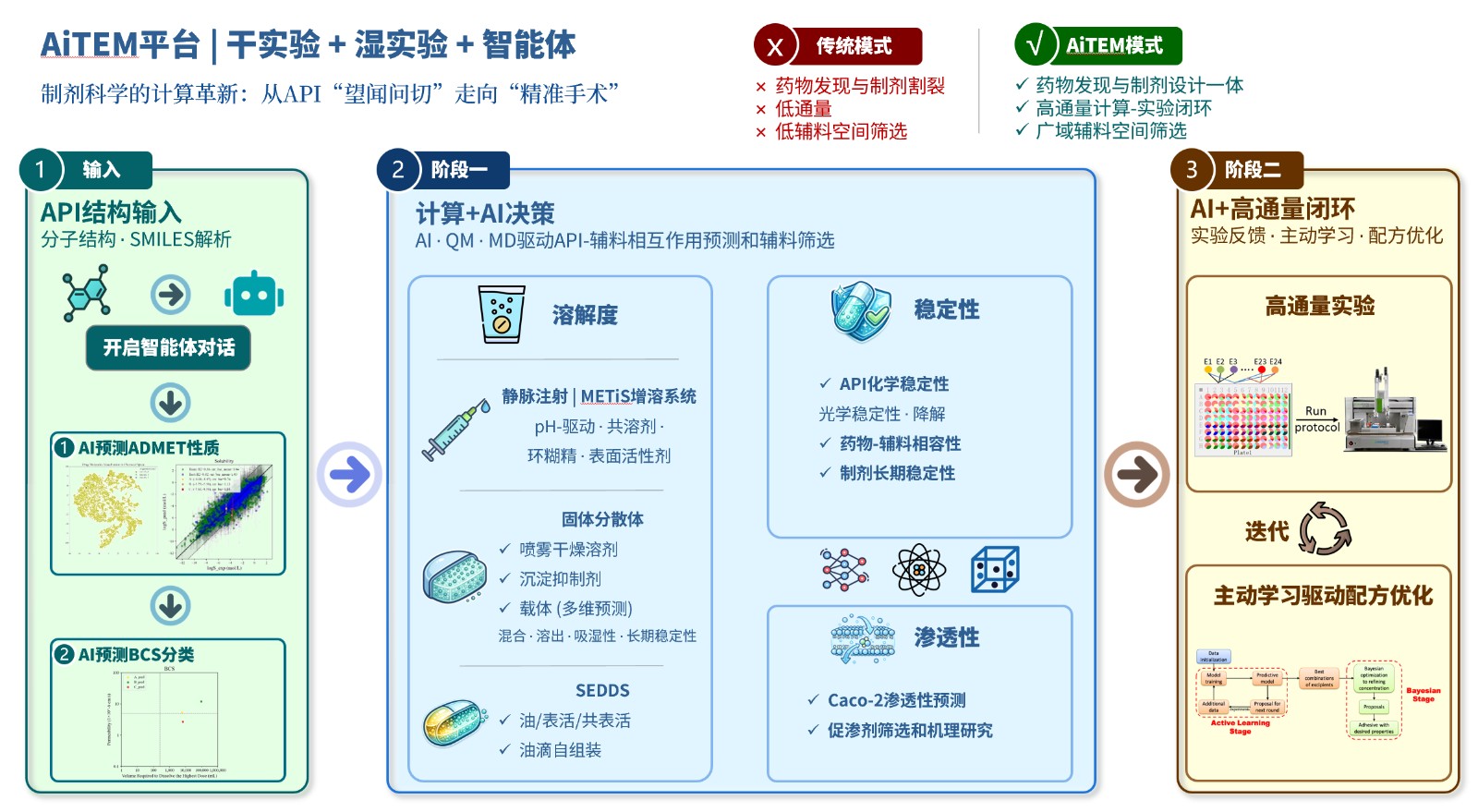 资讯
资讯 剂泰科技与恒瑞医药达成AI制剂合作
8月3日,剂泰科技发布自愿公告,宣布自主研发的AiTEM人工智能制剂创新平台将在恒瑞医药进行本地化部署,加速药物制剂配方开发效率。
2026-08-04 13:01
