
新药审评是美国食品药品管理局(简称FDA)重要职能之一。FDA将加快新药审评、促进创新发展作为其一项重要使命。近年来,FDA开展了一系列改革活动,大大缩短了新药审评时间。从1992年平均需要二年半时间审评一个新药到目前不到十个月审评一个新药,为重症患者获得急需新药赢得了宝贵时间,也为自己树立了一个高效政府机构新形象。
一、FDA简介
FDA是美国历史最悠久的保护消费者组织之一,共约有全职员工15700人,分布在美国各主要城市以及中国、英国、比利时、意大利、印度、墨西哥、智利、哥斯达黎加、南非和约旦等10个国家。其监督管理的产品包括:食品、人用药品、人用生物制品、兽用药品、化妆品、医疗机械、放射性机电产品(包括手机、微波炉等)以及烟草产品,每年监督管理的产品价值超过2万亿美元。2015年度FDA财政预算为47.4亿美元。
FDA局长由美国总统提名并经参议院表决批准。FDA总部划分为五大办公室,分别是局长办公室、食品和兽药办公室、药品和烟草产品办公室、全球监管和政策办公室,以及运行办公室。为履行监管职责,FDA在全国各地设立了许多派出机构。派出机构的总人数约为4500,约占FDA总人数的三分之一。以下重点介绍FDA总部负责新药审评的药品审评与研究中心。
药品审评与研究中心归FDA药品和烟草产品办公室管理。药品审评与研究中心共有4508名员工,划分为12个办公室,分别是:中心主任办公室,对外沟通与交流办公室,法规依从办公室,仿制药办公室,管理办公室,医学政策办公室,新药办公室,药品质量办公室,监管政策办公室,战略项目办公室,监测和流行病学办公室,转化医学办公室。新药办公室是药品审评与研究中心的主要办公室,细分为6个办公室,分别对各类不同的新药进行审评,分别是:抗菌药办公室,负责对抗菌新药的审评;第一药品审评办公室,负责对心血管、肾脏、神经以及精神类疾病新药的审评;第二药品审评办公室,负责对麻醉、止痛、成瘾类、代谢、内分泌、肺、风湿类疾病新药的审评;第三药品审评办公室,负责皮肤、胃肠、先天性代谢缺陷、骨、生殖、泌尿疾病新药的审评;第四药品审评办公室,负责医学影像产品的审评;血液和肿痛药品审评办公室,负责对血液和肿瘤疾病新药的审评。
二、新药审评基本程序
新药审评包括两个过程:一个是新药临床试验申请(简称IND)审评过程,另一个是新药上市申请(简称NDA)审评过程。
(一)IND审评
新药申请人完成新药临床前研究后,就可以向FDA提出IND。如FDA在收到IND之后的30天内未提出反对意见,申请人就可以自行开展新药临床研究。据FDA统计分析,每5 000种进入临床前研究的化合物,仅有5种左右能进入人体实验阶段,最终仅有1种化合物获得上市许可。
IND审评是联系新药临床前研究与临床研究的中间环节。IND审评的目的是确保新药临床研究的安全性,以避免给临床试验受试者带来不必要的风险。IND主要包括两种类型:一种是“商业性IND”,最终目的是为了使新产品获得上市许可,另一种是“非商业性IND”,目的是为了非商业性学术研究,通常由政府机构或科研院所提出这类申请。FDA收到的IND申请大多数属于“非商业性IND”。
IND审评主要侧重于安全性审评。如果FDA对审评结果满意,在收到IND资料后的30天内未做出暂停临床研究的决定,申请人就可以立即开展临床研究。值得注意的是,药品审评与研究中心对IND审评合格的申请并不发出审评合格的通知,故有“没有消息就是好消息”的说法。如果FDA认为IND资料尚存在某些微小缺陷,但不足以作出暂停临床研究的决定,申请人可以在对缺陷项目进行整改的同时开展临床试验。
(二)NDA审评
NDA审评是药品上市前的重要环节。NDA审评的最主要目的是为了确保上市药品的安全有效、质量可控。NDA申报材料主要包括以下15个方面的内容:索引,摘要,化学、生产工艺及质量控制方面数据,样品、包装及标签,非临床药理和毒理数据,人体药代动力学和生物利用度数据,微生物学数据(仅限于抗生素类药品),临床数据,安全性数据更新报告(一般在NDA申报120天后上报),统计学数据,病例报告表,病例报告格式,专利信息,专利声明,其他有关信息。
药品审评与研究中心将申请NDA的新药划分为以下10类:
第1类系指新的分子结构(指从未在美国上市的活性成分);
第2类系指新的活性成分(指已上市产品的新盐结构或新酯结构等);
第3类系指新的剂型;
第4类系指新的组合产品;
第5类系指新的配方或新的生产厂家;
第7类系指已在市场销售但未获得NDA批准的药品;
第8类系指非处方药转换;
第6、9、10类系指新的适应症(针对适应症变化的不同情形)。
药品审评与研究中心将申报的新药按两种审评机制进行审评。一种是正常审评机制;另一种是优先审评机制。
NDA审评主要目的是验证申请人所得出的结论是否正确,药品是否安全有效、质量可控。药品审评与研究中心内部审评人员在审评新药时要展开广泛的交流讨论。例如,医学审评人员在分析临床数据时可能与申请人得出不同的结论,这时医学审评人员就可能与统计学审评人员进行探讨,了解从统计学角度如何看待这个问题。同样,药理学审评人员可能与统计学人员进行密切交流,探讨药品长期动物实验造成潜在致癌性的统计学意义。当药品审评结束时,每个审评员分别写出一份审评报告,交由部门负责人进行汇总分析,并做出是否批准上市的决定。
NDA审评结束时,药品审评与研究中心将根据审评结果,分别做出同意上市、暂不同意上市或不同意上市三种决定。收到同意上市决定的产品可立即在美国上市,收到暂不同意上市决定的产品需要就某些细节问题进行修改或补充有关资料,收到不同意上市决定的产品需进行较大范围的变更或补充资料。一般来说,产品上市许可决定由处长签发,但第1类产品上市许可决定需要更高一级的办公室负责人签发。例如,药品审评与研究中心的第一药品审评办公室下设3个处,分别是心血管、肾脏类产品处,神经类产品处,以及精神类产品处。通常都是这3位处长负责签发新药上市许可决定,只有第1类产品上市许可决定由第一药品审评办公室主任签发。
三、FDA加快新药审评政策
美国、欧洲和日本是人用药品注册技术规定国际协调会议(ICH)的创始成员单位,在药品注册审评方面采用许多相同或相似的技术指南,但在新药审评速度方面,美国却明显快于欧洲和日本,很多全球创新性新药率先在美国获得上市许可。2013年美国FDA共批准27个新药,其中20个新药系全球首次上市的新药,首次上市的比例高达74%。
美国《处方药用户付费法案》规定,对属于优先审评的创新药,美国FDA应在6个月内完成百分之九十的品种审评。对属于正常审评的创新药,美国FDA应在10个月内完成百分之九十的品种审评。2014年美国FDA共批准41个创新性新药,其中40个新药在6个月或10个月内完成审评。
FDA主要有四种新药加快审批途径,分别是快速通道、突破性疗法、加速批准和优先审评。快速通道针对的是治疗严重疾病且目前临床用药空缺的新药,突破性疗法针对的是对现有疗法有明显改善的新药,加速批准针对的是目前临床用药空缺、基于替代终点而批准的新药,优先审评是指在申请受理后6个月内完成审评的新药。
这四种途径的主要异同点如下:

尽管四种快速审评途径的评判标准各不相同,但实际上四种快速途径之间存在相似性,一种新药往往可以获得不止一种快速审评通道。例如,2014年FDA共批准41个新药,其中纳入快速通道的新药17个,占41%,纳入突破性疗法通道的新药9个,点22%,纳入加速批准通道的新药8个,占22%,纳入优先审评通道的25个,占61%。在41个新药当中,其中27个新药获得不止一种快速审评通道,比例高达66%。
四、美国加快新药审评最新立法进展
尽管美国新药审评速度遥遥领先于其他国家,但美国并不因此故步自封。2015年7月10日,美国众议院以344票赞成、77票反对的投票结果轻松通过了《21世纪治疗法案》(21st Century Cures Act)。该法案长达362页,其核心是加快新药研究、开发和审评。目前该法案正在美国参议院审议之中。
尽管《21世纪治疗法案》能否最终通过参议院表决,目前还不得而知,但从该法案在众议院获得的广泛支持来看,参议院通过该法案的可能性较大。法案有关药品审评的主要内容包括:
(一)准许FDA提高员工工资待遇,以帮助FDA招聘和留住优秀高端人材。以前美国法律规定,FDA的高级雇员人数不得超过500名,工资待遇不得超过行政I级。2015年美国行政I级的年收入为203700美元。美国国务卿等21名高官为行政I级待遇。新法案规定FDA的高级雇员人数将不受限制,只要符合任职条件就可以聘任为高级雇员;同时,准许优秀高端人材的工资待遇超过行政I级,只有不超过美国总统年薪即可。目前美国总统年薪为40万美元。法案希望通过此举将流向药品生产企业或医疗器械生产企业的高端人材吸引到FDA任职。
(二)增加国立卫生研究院和FDA年度预算。医药基础研究是发现新药的重要源泉。基础研究取得突破往往能带动一批新药产品的开发。据分析,该法案之所以能高票在众议院获得通过,很重要一个原因是国会议员们高度赞同法案中关于给美国国立卫生研究院(简称NIH)增加年度预算的条款。2015财年NIH的预算为303亿美元。如法案获得通过,2016、2017、2018财年NIH预算将分别增加到318亿美元、333美元、348亿美元。另外,NIH还将新设立创新基金,鼓励医学前沿学科研究和优秀青年科学家创新。未来5年FDA也将新获得5.5亿美元拨款,用于药品监管科学研究。
(三)鼓励FDA药品审评制度改革,将患者意见和建议更多纳入药品审评决定中。美国国会认为,没有人比患者更能理解疾病所带来的痛苦,也没有人比患者更有资格权衡某一药品的风险和益处。因此药品审评过程中应该充分考虑患者的意见和建议。
(四)鼓励FDA进一步研究加快药品审评的手段,如扩大生物标记物的应用等。
(五)加快抗菌药的研发,允许仅经过小范围临床试验就批准某些抗菌药。
(六)准许具有重大创新的医疗器械产品纳入FDA加快审评通道。
(七)将罕见病的新适应症独占期延长6个月。
来源:商务部

为你推荐
 资讯
资讯 悦唯医疗完成近亿元A++轮投资,加速重症冠心病诊疗全流程创新器械研发与国产替代
此次融资将主要用于深化冠心病诊疗全流程创新器械和脉动式左心室辅助系统等新产品的研发,以及加速已获准上市的心脏稳定器等产品的市场推广。
2025-04-03 09:28
 资讯
资讯 海尔盈康一生启动孤独症儿童关爱行动,创新罕见病可持续公益新生态
本次活动聚焦孤独症儿童的诊疗,探讨交流AI赋能全流程防治康体系创新、前沿性生物科技诊疗技术等话题,旨在通过生态联盟的力量推动医学研究、科技创新与人文关怀的融合,让“星...
2025-04-03 09:11

 资讯
资讯 《NPJ digital medicine》刊发李冬梅教授团队成果:AI赋能高效识别眼睑肿物
亚太眼整形外科学会主席、中华医学会眼科分会眼整形眼眶病学组副组长李冬梅教授团队携手爱尔数字眼科研究所,在《NPJ digital medicine》(影响因子:12 4)学术期刊发表团队...
文/李林 2025-04-02 10:27

 资讯
资讯 默克全球执行副总裁周虹:合作与创新是默克未来五年战略的两大关键词
近日,德国默克医药健康全球执行副总裁、中国及国际市场负责人周虹带领医药健康中国及国际市场管理团队开启了2025年度首次“中国行”。
2025-04-01 17:11
 资讯
资讯 首个且唯一,阿斯利康PD-L1单抗获FDA批准治疗肌层浸润性膀胱癌
度伐利尤单抗联合吉西他滨和顺铂作为新辅助治疗,随后度伐利尤单抗作为根治性膀胱切除术后的辅助单药治疗,用于治疗肌层浸润性膀胱癌成年患者。
2025-04-01 14:37
 资讯
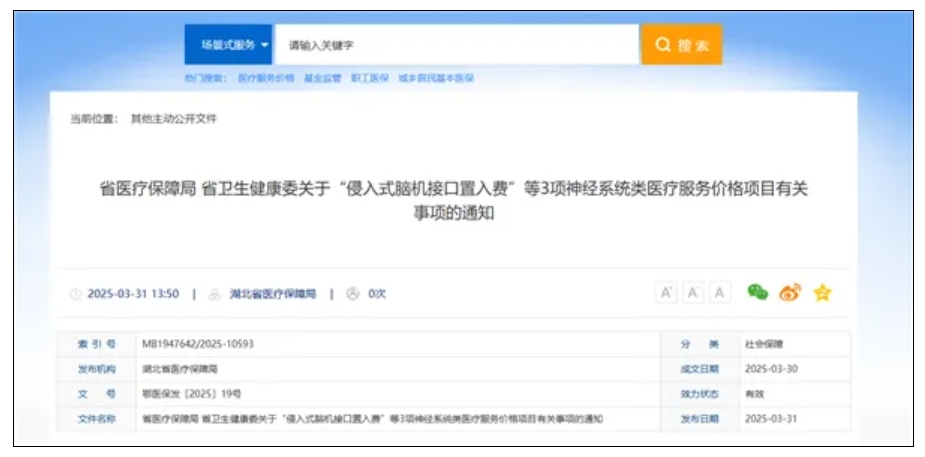
资讯 全国首个,湖北为脑机接口医疗服务定价
昨日(3月31日),据“湖北发布”消息,湖北省医保局发布全国首个脑机接口医疗服务价格,其中,侵入式脑机接口置入费6552元 次,侵入式脑机接口取出费3139元 次,非侵入式脑机...
2025-04-01 11:03
 资讯
资讯 一款国产创新流感药,获批
近日,据国家药监局官网信息显示,青峰医药下属子公司江西科睿药自主研发的1类创新药玛舒拉沙韦片(商品名:伊速达)正式获批上市,用于既往健康的12岁及以上青少年和成人单纯性...
2025-04-01 10:22
 资讯
资讯 26省联盟药品集采启动,聚焦妇科用药和造影剂
近日,山西省药械集中招标采购中心发布《关于做好二十六省联盟药品集中带量采购品种数据填报工作的通知》,开展相关采购数据填报工作。
2025-03-31 21:48
 资讯
资讯 优时比罗泽利昔珠单抗注射液(优迪革)中国获批,全球首个且唯一双亚型创新药治疗全身型重症肌无力
作为唯一人源化、高亲和力且具备创新修饰结构的IgG4单抗,关键Ⅲ期MycarinG试验证实罗泽利昔珠单抗注射液(优迪革®)较安慰剂显著改善全身型重症肌无力患者的多个临床终点与结局。
2025-03-31 15:58
 资讯
资讯 从手术麻醉到生命全周期护航,麻醉学科发展拓宽生命边界
3月26日,由中华医学会麻醉学分会、中国医师协会麻醉学医师分会等23家学协会共同举办的2025年中国麻醉周学术活动的启动仪式举办,该活动以“生命之重,大医精诚——守生命保驾护...
2025-03-31 15:30
 资讯
资讯 欧狄沃联合逸沃成为中国目前唯一获批的肝细胞癌一线双免疫联合疗法
欧狄沃联合逸沃对比仑伐替尼或索拉非尼,可显著改善不可切除肝细胞癌一线患者的总生存期(OS),客观缓解率(ORR)可改善近3倍,中位缓解持续时间(mDOR)达30个月
2025-03-31 13:45
 资讯
资讯 罗氏制药榜首 “现金牛” 产品罗可适(奥瑞利珠单抗)在华获批:开启多发性硬化症一年两次治疗新时代
罗氏制药今日(3月31日)宣布,其旗下创新药罗可适®(Ocrevus®,通用名:奥瑞利珠单抗注射液 ocrelizumab injection)正式获得中国国家药品监督管理局批准,每六个月静脉输...
2025-03-31 13:39
 资讯
资讯 三生有幸,医者仁心:三生制药向全体医药工作者致敬!
3月30日是国际医师节,由三生制药公益支持的以“三生有幸,医者仁心”为主题的公益活动,携手20位医生代表,以寄语海报的形式,共同向全体医护人员表达诚挚的祝福与关爱。
2025-03-30 17:38
 资讯
资讯 新版药典自2025年10月1日起实施
3月25日,国家药监局官网发布《国家药监局 国家卫生健康委关于颁布2025年版的公告(2025年第29号)》,2025年版《中国药典》自2025年10月1日起施行。
2025-03-30 17:07