
5月17日,在2016中国国际药物信息大会暨第八届DIA中国年会上,CFDA药化注册司副司长李茂忠就当前仿制药一致性评价最受关注问题之一“愿意承接一致性评价的临床机构数量有限,首批开展品种在2018年底完成困难”作答:“CFDA此前公示的首批开展一致性评价工作的截止日期不会改变,我们将在实际工作中具体了解机构对BE试验的承接情况;同时,CFDA将与卫计委联动,积极推动问题解决”。
临床资源到底是否足够?
Insight数据显示,首批需要在2018年底前完成一致性评价共涉及292个基本药物目录品种,共涉及19715个批准文号,其中可以确定的有效批文为70.34%;涉及企业数量为2028家,其中7家企业批文数量超过100个,47家企业所持批文数量为50-100个,529家企业批文数量在10-50之间;上海医药、华润双鹤、白云山、哈药、复星医药五家上市公司分别以505、211、200、144、132个批文数量位列所持批准文号数量排行榜前五位。
目前,CFDA仍有数项关于此次仿制药一致性评价工作的具体要求和指导原则处于征求意见阶段,随着正式文件的陆续发布,留给企业实际操作的时间也越来越短。而国务院办公厅在2016年3月5日发布的《关于开展仿制药质量和疗效一致性评价的意见》中强调,逾期未完成的,不予再注册。
截止2015年底,全国共有433家GCP证书未到期,而根据CDE临床试验登记平台和中国临床试验注册中心ChiCTR数据,只有122家开展过BE/I期项目,而在2015年9月CFDA发布的核查清单中,82家机构涉及“722自查核查风暴”。此前2016年1月,业界曾有一项调研显示,400余家临床试验机构中仅有53家机构表示愿意承接BE/I期项目。
做一个简单的计算,数据预测基于不完全的业界数据和经验,如有异议敬请留言:
暂不考虑BE豁免。
假如,拥有24张床位的临床试验机构平均开展一项临床试验的平均时间为1个月;
假如,CFDA关于此次仿制药一致性评价的所有工作要求能够在今年6月前全部发布,那么距离2018年底,还有30个月的时间;
假如,上述愿意承接BE/I期53家机构平均有24张BE/I期床位;
推导一下,只有30*53=1590个品种来得及通过这一轮仿制药一致性评价,每个品种平均有5.4个批文可以获得BE资源。
这意味着,在规定时限内,逾13800个批准文号中的近90%都将没有临床资源可用。对企业来说,无论产品此前销售额如何,谁能抢占到BE资源,谁将成为主宰2019年以后市场的、最幸运的5.4个品种之一。
这或许也是,近期频繁被爆出的、高达300万-500万元的BE临床试验费用的缘由。
然而对药企来说,这不仅增加以“价廉”为特征的基本药物现有成本,未来还可能造成更多的基本药物短缺问题;此外,CFDA在临床试验质量自查核查以来一直强调的“由企业承担主体责任”理念也将面临挑战:面对一个有合法伤害权的权利怪兽,企业如何履行监察的职能?
“我们要努力营造一个‘良药走遍天下,劣药寸步难行’的社会环境”。这是CFDA局长毕井泉在上任伊始在北京大学演讲时传递的信念。这意味着,给良药以机会,而不是让良药在自证之前先掉层皮。
如此局面,如何破?
需要注明的是,上述论证是在仅有53家机构愿意承接BE试验的前提下才会产生的困境。如果曾经接开展过BE/I期临床试验的122家机构都参与进来,临床资源将相对充裕;如果强制全部拥有GCP证书的机构都承接BE试验项目,前述过万的有效批文都将不存在BE试验资源紧缺的情况。
然而,很多临床试验机构不愿承担BE/I期临床试验的情况客观存在,其理由包括但不限于此类试验并不能够帮助所在临床研究机构建立声誉、帮助相关研究者建立学术地位。因此,破解临床试验资源优先困境不仅需要CFDA尽快组织情况摸底、重新审视GCP认证,也需要卫计委协调实际资源。
据记者了解,自2015年12月国务院同意建立药品医疗器械审评审批制度改革部际联席会议制度,由CFDA牵头、卫计委参与的部际联席会议已成功召开一次。而截止目前,关于临床试验资源配置的议题尚未进入部际联席会议的讨论范围。
医谷链
来源:E药经理人 作者:马茗舒

为你推荐

 资讯
资讯 带状疱疹疫苗“遇冷”,百克生物2024年净利润腰斩
近日,国内疫苗龙头企业百克生物发布2024年年报,数据显示,其报告期内实现营收12 29亿元,同比下降32 64%;归属于上市公司股东的净利润2 32亿元,同比下降53 67%。对于营收...
2025-04-23 12:59





 资讯
资讯 重庆常用药联盟接续集采中选结果
近日,重庆常用药联盟接续集采中选情况公布,该联盟由重庆牵头,联合湖北、广西、海南、贵州、云南、青海、宁夏、新疆及新疆生产建设兵团等十省(区、市)开展的药品集中带量采...
2025-04-21 18:48

 资讯
资讯 全周期智控慢病,诺和诺德与京东健康开启战略合作
2025年4月21日,全球领先的生物制药公司诺和诺德与京东健康在北京正式签署战略合作协议,标志着双方在糖尿病和体重管理领域的合作进入新阶段。依托诺和诺德百年深耕慢病领域的专...
2025-04-21 15:57
 资讯
资讯 康方生物1类新药依若奇单抗上市申请获批,用于中重度斑块状银屑病成人患者
该药是我国第一个且唯一获批上市的IL-12 IL-23“双靶向”单克隆抗体新药,是康方生物自身免疫性疾病领域首个获批上市的一类新药。
2025-04-21 13:39
 资讯
资讯 阿斯利康乳腺癌1类创新药卡匹色替片中国获批
该药适用于联合氟维司群用于转移性阶段至少接受过一种内分泌治疗后疾病进展,或在辅助治疗期间或完成辅助治疗后12个月内复发的激素受体(HR)阳性、人表皮生长因子受体2(HER2)...
2025-04-21 11:02
 资讯
资讯 辉瑞宣布终止一款口服GLP-1减肥药的临床开发
近日,辉瑞在其官网宣布,决定终止开发口服胰高血糖素样肽-1受体(GLP-1R)激动剂Danuglipron(PF-06882961),原因系在一项有关用药剂量的临床试验中,一名患者出现了可能由该...
2025-04-21 10:29
 资讯
资讯 福建省医保局印发单列门诊统筹支付医保药品目录(2024年版)
根据2024年6月发布的《福建省医保药品单列门诊统筹支付管理办法(试行)》,为了让参保患者无需住院、在门诊就医也能用上国家谈判药品、享受医保待遇,将适用于门诊治疗、使用周...
2025-04-20 13:34
 资讯
资讯 首批中国消费名品名单,医药健康企业有哪些?
近日,工业和信息化部办公厅发布首批中国消费名品名单,分为中国消费名品名单和中国消费名品成长企业名单。首批中国消费名品名单共包括93个企业品牌和43个区域品牌。中国消费名...
2025-04-20 11:17
 资讯
资讯 携手共绘“个性化近视手术”新蓝图:爱尔眼科与爱尔康启动100家医院全光塑技术战略合作
双方将以技术共享为核心,以人才培养为支撑,以科研协作为纽带,全力推进屈光手术标准化诊疗体系建设,加速前沿技术在临床领域的普及应用
文/ 屈慧莹 2025-04-19 23:35
 资讯
资讯 CDE:简化港澳已上市传统口服中成药内地上市注册审批申报资料及技术要求
允许香港、澳门特区本地登记的生产企业持有,并经香港、澳门特区药品监督管理部门批准上市且在香港、澳门特区使用15年以上,生产过程符合药品生产质量管理规范(GMP)要求的传统...
2025-04-18 18:54
 资讯
资讯 君德医药完成近亿元A轮融资,加速推进创新药械组合平台建设与产品上市
本轮融资主要用于首个减重口服器械的注册及生产销售,以及加速多个核心创新药械组合技术平台的产品管线研发进程。
2025-04-18 14:34
 资讯
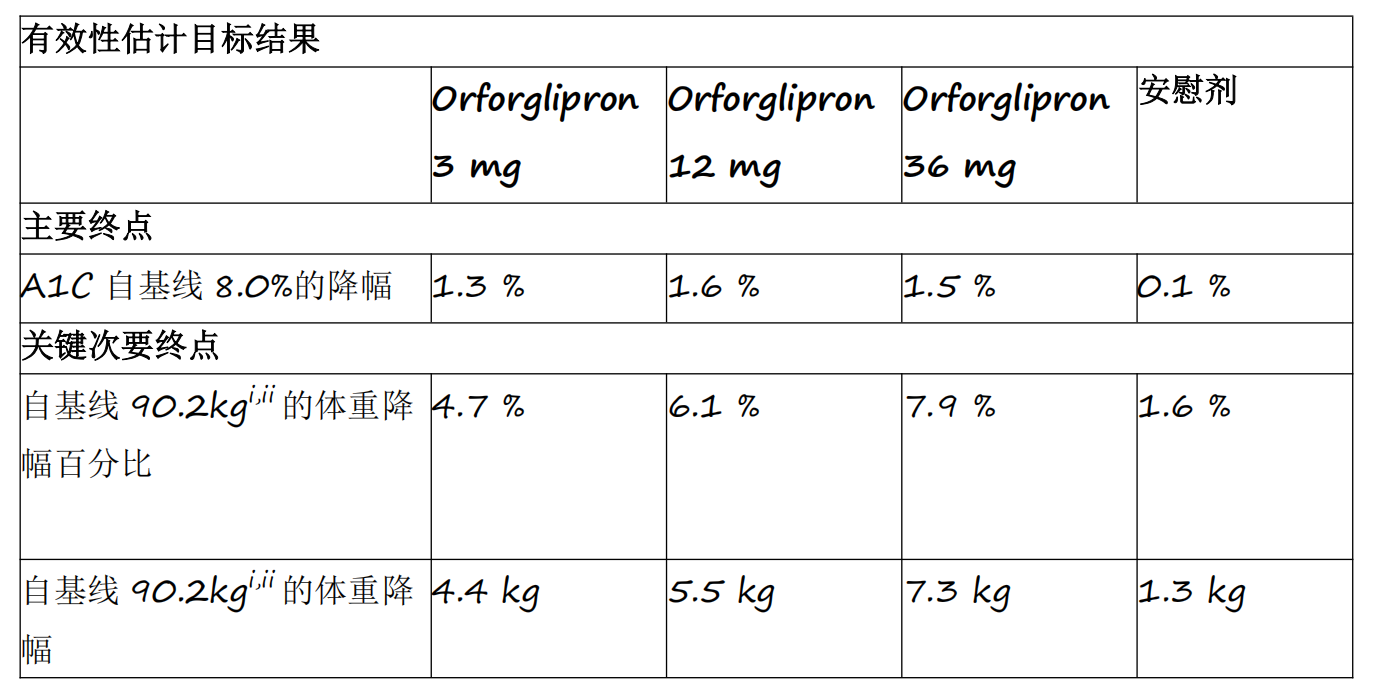
资讯 礼来首个小分子口服GLP-1RA药物orforglipron 3期临床研究成功
Orforglipron是首个成功完成3期临床研究的小分子GLP-1类药物,各剂量组平均A1C降幅为1 3%至1 6%
2025-04-18 14:12