
今日(7月5日),为加强医疗器械临床试验监督管理,进一步提高注册审查质量,国家药品监督管理局组织制定了《角膜塑形用硬性透气接触镜临床试验指导原则》《软性接触镜临床试验指导原则》,具体内容如下:
附件:1.角膜塑形用硬性透气接触镜临床试验指导原则
为进一步规范角膜塑形用硬性透气接触镜(以下简称角膜塑形镜)产品上市前的临床试验,并指导该类产品申请人在申请产品注册时临床试验资料的准备,根据《医疗器械临床试验质量管理规范》,制订本临床试验指导原则。
随着角膜塑形镜产品以及眼科学诊疗技术的发展和相关法规政策、标准制定等情况的变化,本指导原则将会不断地完善和修订。
一、适用范围
结合法规的具体要求,要求其进行完整的上市前临床试验时,适用于本项指导原则。
本指导原则适用于采用塑形方法暂时改变角膜形态,达到暂时性矫正屈光不正为预期目的的硬性透气接触镜产品的临床试验。
特殊设计的角膜塑形镜产品须结合申报产品自身特点另行制定其上市前临床试验方案,原则上不应低于本项指导原则的要求。
二、基本原则
申请人应当按照《医疗器械临床试验质量管理规范》的相关要求设计、实施、监查、记录、总结临床试验结果,并保证临床试验过程规范,结果真实、科学、可靠和可追溯。
三、临床试验方案
(一)临床试验目的及注意事项
角膜塑形镜产品的临床试验目的是评价申报产品是否具有预期的安全性和有效性。在临床试验中推荐使用LogMAR视力表(也称为EDTRS视力表),也可使用标准对数视力表。应采用标准的方法检查视力。如为多中心临床试验,不同的临床试验机构中使用的视力表须一致。同时涉及屈光度检查时,均应以受试者主觉验光值(包括球镜度及柱镜度)为准,客观验光数值(包括球镜度及柱镜度)应记录并作为参考。
(二)临床试验设计
以申请角膜塑形镜上市为目的的临床试验应是前瞻性、随机对照临床试验,临床试验应当在两个或者两个以上医疗器械临床试验机构中进行,如按照同一临床试验方案在3个以上(含3个)临床试验机构实施的临床试验将视为多中心临床试验。对照用医疗器械应选择中国已经批准上市的同类产品,其主要功能原理必须与受试产品一致,配戴方式必须一致,不建议采用历史对照或非平行对照。
(三)临床试验样本量
主要评价指标:30天的产品有效率。有效定义:当裸眼视力及屈光度均达到如下临床评价要求时为“有效”:
1.裸眼视力:裸眼视力应大于等于0.8。
2.残余屈光度:残余屈光度应小于±0.50D。
根据对照用角膜塑形镜的相应指标的循证医学相关资料做出检验假设并计算样本量。同时,样本量的确定与选择的假设检验类型(优效、非劣效、等效性检验)及I、II类错误和具有临床意义的界值(疗效差)有关,同时还应考虑预计排除及临床失访的病例数。
临床试验样本量的确定应当符合临床试验的目的和统计学要求,并且完成所有访视的受试者不应少于本指导原则中规定的最低样本量。目前,角膜塑形镜随机对照试验为与对照产品进行的1:1的不少于100对(200个受试者)的临床试验,要求受试者双眼均入组观察,双眼数据均须纳入统计分析,不推荐单眼患者入组。
(四)临床试验随访时间
不同产品的临床试验随访时间不完全一致,随访时间的确定应该具有医学文献资料支持,要有医学共识。
目前,角膜塑形镜的临床试验随访时间至少为12个月。同时,应当科学设置访视时间点(至少应包含戴镜后1天、1周、2周、30天、3个月、6个月、9个月、12个月)。
(五)临床试验受试者的入选标准及退出标准
临床试验受试者的入选标准应当考虑申报产品的适用范围。入组过程中,应在遵循随机原则的基础上,尽量兼顾组内及组间均衡性,可以包括但不限于:
1.近视度数为4.00D或更低。
2.角膜曲率计及角膜地形图。
3.柱镜度,顺规散光应低于1.75D,逆规散光应低于1.00D;
4.年龄(应为实足年龄)。应分为年龄≤13岁、13岁<年龄<18岁、年龄≥18岁共3组,研究对象应在3组间均衡分布,年龄≤13岁、13岁<年龄<18岁组每组不小于30例。
5.治疗前屈光度应稳定。
6.无角膜屈光手术病史。
7.眼球尤其是角膜应健康。
8.全身状况健康。
9.未使用影响眼球及角膜的药物。
10.女性未妊娠,且近期无妊娠计划。
制定受试者退出标准,统计受试者退出人数及原因。
(六)临床试验的有效性指标
临床试验的有效性指标在每次临床访视中均须如实记录。临床试验的有效性指标包括但不限于:
1.裸眼视力。应至少列出在戴镜后1天、1周、2周、30天、3个月、6个月、9个月、12个月及最终时间点裸眼视力。提供裸眼视力在试验组及对照组的统计学分析结果。
2.屈光度。应至少列出在戴镜后1天、1周、2周、30天、3个月、6个月、9个月、12个月及最终时间点的屈光度。提供屈光度在试验组及对照组的统计学分析结果。
3.角膜地形图。应至少列出在戴镜后1天、1周、2周、30天、3个月、6个月、9个月、12个月及最终时间点的角膜地形图的相关重要参数,提供试验组及对照组的统计学分析结果。
(七)临床试验的安全性指标
临床试验的安全性指标包括但不限于:
1.症状、体征、并发症、不良事件等。推荐在戴镜后1天、1周、2周、30天、3个月、6个月、9个月、12个月及最终时间点列出受试者的症状、体征、并发症、不良事件等,需提供试验组及对照组的统计学分析结果。
2.角膜曲率计及角膜地形图。推荐在戴镜后1天、1周、2周、30天、3个月、6个月、9个月、12个月及最终时间点的列出受试者的角膜曲率计及角膜地形图相关重要参数,需提供试验组及对照组的统计学分析结果。
3.角膜厚度及角膜内皮细胞参数。推荐在基线、研究中和末次访视时测量受试者的角膜厚度及角膜内皮细胞数,应分析试验组及对照组角膜塑形镜对角膜厚度及角膜内皮细胞数的影响。
4.最佳矫正视力。应至少列出在戴镜后1天、1周、2周、30天、3个月、6个月、9个月、12个月及最终时间点的访视时最佳矫正视力和初始最佳矫正视力分析结果,比较试验组及对照组中与初始最佳矫正视力相比最佳矫正视力下降1行、2行、或2行以上的受试者的比率。
5.柱镜度。应至少列出在戴镜后1天、1周、2周、30天、3个月、6个月、9个月、12个月及最终时间点的访视时柱镜度的分析结果,比较试验组及对照组中与初始屈光度相比,柱镜度增加1.00D以下、1.00D至2.00D、2.00D以上受试者的比率。
6.眼内压。推荐在戴镜后基线、研究中和末次访视时测量受试者的眼内压,应分析试验组及对照组角膜塑形镜对眼内压的影响。
7.角膜塑形镜的配适状态。应至少列出在戴镜后1天、1周、2周、30天、3个月、6个月、9个月、12个月及最终时间点的访视时角膜塑形镜的适配状态。
8.记录镜片的破损率、划痕、蛋白沉淀等情况。应至少列出在戴镜后1天、1周、2周、30天、3个月、6个月、9个月、12个月及最终时间点的访视时镜片的破损率、划痕、蛋白沉淀等情况。
(八)统计分析方法
数据分析时应考虑数据的完整性,所有签署知情同意并使用了受试产品的受试者必须纳入最终的统计分析,应提供患者水平(受试者数)及病例水平(受试眼数)的主要疗效指标分析结果。数据的剔除或偏倚数据的处理必须有科学依据和详细说明。
临床试验的数据分析应基于不同的分析集,通常包括全分析集(Full Analysis Set,FAS)和符合方案集(Per Protocol Set,PPS),研究方案中应明确各分析集的定义。主要评价指标的分析应同时在全分析集和符合方案集上进行,以评价结果的稳定性。全分析集中脱落病例,其主要评价指标缺失值的填补方法应在方案中予以事先说明,并于研究结束后进行灵敏度分析,以评价缺失数据对研究结果稳定性的影响。
临床试验数据的分析应采用国内外公认的经典统计分析方法。临床试验方案应该明确统计检验的类型、检验假设、判定疗效有临床意义的界值等,界值的确定应有依据。
对于主要评价指标,统计结果需采用点估计及相应的95%置信区间进行评价。不能仅将p值作为主要评价指标的评价依据。
(九)临床试验报告和统计分析报告
1.临床试验报告。由临床试验牵头单位根据基于所有入选受试者的总的统计分析报告,出具临床试验报告。各临床试验单位出具临床试验小结。各临床试验单位不需要单独出具分中心统计报告。临床试验报告内容包括:试验目的、试验假设、主要评价指标、评价方法、对照品、入选/排除标准、样本量及计算依据、受试者资料、试验质量控制措施、数据管理及质控措施、试验结果、伴随治疗、不良事件、并发症及其处理、试验结论、适用范围、禁忌症和注意事项、存在问题及改进意见等。
此外,需注意以下问题:
(1)临床试验报告应与临床试验方案保持一致。
(2)明确所有病例是否全部完成随访,所有接受了医疗器械治疗的病例是否均纳入最终的统计分析,失访病例需明确失访原因。
(3)提交疗效评价与安全性评价统计过程中所涉及的原始数据。
(4)报告所有不良事件发生的时间、原因、后果及与试验用器械的关系,对于所采取的处理措施需予以明确。
2.统计分析报告。应将所有中心的数据合并在一起进行统计分析,并出具总的统计分析报告。应对随机对照部分和单组部分数据分别进行统计分析,并出具相应的统计分析报告。为了保证受试者的安全性和数据的完整性,建议采用中央注册或中央随机系统分配治疗。应对所有入选的受试者进行数据管理和质量控制,遇有不清楚的问题时,应通过临床试验的监查员与原始记录核对。统计分析报告应至少包括如下4部分内容:
(1)临床试验完成情况描述:包括临床试验概况(筛选人数、入组人数、完成试验人数、失访/退出/剔除人数等);
(2)基线描述:应对所有入选受试者(ITT分析集)的基线人口统计学指标、生命体征及其他相关病史指标等进行描述;
(3)疗效/效果评价:应对全分析集和符合方案集分别进行统计分析;
(4)安全性评价时,应对所有入组的受试者进行分析,不能遗漏所有发生的任何不良事件。同时,详细描述每一病例出现的全部不良事件的具体表现、程度、预后及其与研究产品的关系。
四、参考文献:
1.《医疗器械注册管理办法》(国家食品药品监督管理总局令第4号)
2.《医疗器械临床试验质量管理规范》(国家食品药品监督管理总局 中华人民共和国国家卫生和计生委员会令第25号)
3.《关于发布医疗器械临床评价技术指导原则的通告》(国家食品药品监督管理总局通告2015年第14号)
4.Guidance for Premarket Submissions of Orthokeratology Rigid Gas Permeable Contact Lenses. (美国FDA)
5.瞿佳。《视光学理论和方法》人民卫生出版社 2004
6.葛坚。《眼科学》人民卫生出版社 2005
五、起草单位
本指导原则由国家食品药品监督管理总局医疗器械技术审评中心起草并负责解释。
附件2.软性接触镜临床试验指导原则
随着科学技术的不断发展,软性接触镜产品日益增多。为了进一步规范该类产品上市前的临床试验,并指导该类产品申请者/生产企业在申请产品注册时临床试验资料的准备,特制订本指导原则。
本指导原则虽然为该类产品的临床试验及申请者/生产企业在申请产品注册时临床试验资料的准备提供了初步指导和建议,但是不会限制医疗器械相关管理部门对该类产品的技术审评、行政审批以及申请者/生产企业对该类产品临床试验资料的准备工作。
随着软性接触镜材料的进步以及眼科相关诊治技术的发展、相关法规政策和标准的更新,本指导原则还会不断地完善和修订。
一、适用范围
按照《医疗器械注册管理办法》规定需在国内进行上市前临床试验的日戴软性接触镜注册申报项目,可参考本指导原则制定临床试验方案并实施。
本指导原则适用于采用光学成像原理、矫正屈光不正的日戴软性接触镜(不含采用柱镜设计针对散光进行矫正的软性接触镜)。特殊功能设计的软性接触镜产品须结合申报产品自身特点另行制定其上市前临床试验方案。
二、基本原则
软性接触镜的临床试验应符合《医疗器械临床试验质量管理规范》及其他相关法律、法规的规定。
进行上市前临床试验的软性接触镜应已经过相对科学的实验室研究和动物实验验证,且研究结果可基本证明产品安全、有效。
三、临床试验方案
(一)临床试验目的及注意事项
软性接触镜产品的临床试验目的是评价申报产品是否具有预期的临床安全性和有效性。在临床试验中推荐使用标准对数视力表,也可使用标准对数视力表。应采用标准的方法检查视力。如为多中心临床试验,不同的临床试验机构中使用的视力表须一致。同时涉及屈光度检查时,均应以受试者主觉验光值(包括球镜度及柱镜度)为准,客观验光数值(包括球镜度及柱镜度)应记录并作为参考。
因临床方案是阐明试验的总体设计、试验目的、试验方法和步骤等内容的文件,应当最大限度地保障受试者的安全和健康,应当由申办者和研究者按规定的格式共同设计制定,报伦理委员会批准后实施,若有修改,必须重新经伦理委员会批准。
(二)临床试验设计
以申请软性接触镜上市为目的的临床试验应是前瞻性、随机对照临床试验,临床试验应当在两个或者两个以上医疗器械临床试验机构中进行,如按照同一临床试验方案在3个以上(含3个)临床试验机构实施的临床试验将视为多中心临床试验。对照用医疗器械应选择境内已经批准上市的同类产品,应考虑其主要功能原理与受试产品一致,配戴方式一致,配戴周期相同,镜片材料相似,不建议采用历史对照或非平行对照。
(三)临床试验样本量
主要评价指标:戴镜有效率。有效定义:戴镜一周时分别检查两眼矫正视力均大于等于5.0(标准对数视力表)为“有效”。
根据对照用软性接触镜的相应指标的循证医学相关资料做出检验假设并计算样本量。同时,样本量的确定与选择的假设检验类型(优效、非劣效、等效性检验)及I、II类错误和具有临床意义的界值(疗效差)有关,同时还应考虑预计排除及临床失访的病例数。
临床试验样本量的确定应当符合临床试验的目的和统计学要求,并且完成所有访视的受试者不应少于本指导原则中规定的最低样本量。目前,软性接触镜产品的有效性、安全性的评估均应采用临床上通用的评价标准,每个评价病例应该是完整的双眼数据,临床试验最终完成总样本量不少于120例,非劣效界值不大于10%,试验组不少于60例。
(四)临床试验随访时间
不同产品的临床试验随访时间不完全一致,随访时间的确定应该具有医学文献资料支持,要有医学共识。
目前,软性接触镜的临床试验至少应分别于戴镜当天和戴镜1周、1个月、3个月进行随访观察。(可结合试验产品的临床观察具体情况设定更为频繁的观察时间)
(五)入选标准
试验组和对照组需采用统一的入选标准和排除标准,要求受试者分别检查两眼,框架镜最佳矫正视力均能达到5.0。其他入选标准和排除标准的具体内容由临床试验负责单位具体讨论决定。
(六)临床观察指标
1.有效性评价项目。
视力:框架镜最佳矫正视力、接触镜矫正视力
可使用我国标准对数视力表检查受试者的框架镜最佳矫正视力和接触镜矫正视力(远用)并记录。
2.安全性评价项目。
(1)眼部情况:在临床试验期间要求定期随访观察眼部的变化,包括:裸眼视力(远用)、眼睑、睑缘、泪液膜、结膜、角膜、前房、虹膜、瞳孔、晶状体、眼底、眼压等,在临床观察期间需严密监控并记录临床并发症的发生,随访次数由申办者与研究者在研究方案中确定,原则上不能少于3次。
(2)镜片配适状态:初次配戴及定期随访中需观察镜片在眼表的位置(中心定位)、覆盖度、松紧度、活动度,并评价和记录其等级。
(3)镜片状态:在定期随访中观察镜片的污损情况如:沉淀、变形、变色、锈斑、划痕、破损等。
(4)屈光状态:屈光度和角膜曲率,应提供受试者戴镜前、后的屈光度变化(球镜度及柱镜度)和角膜曲率变化。
目前软性接触镜的临床常规疗效评价指标可参考附录Ⅰ和附录Ⅱ
(七)确定护理系统
根据受试产品的特性选择适合的并已经在我国批准上市的护理产品,要求试验组与对照组使用相同的护理用品。
在定期随访中同时观察镜盒中护理液和瓶中护理液有无混浊、杂质、沉淀及对眼睛的刺激症状等。
(八)临床随访观察内容
在随访中对软性接触镜产品的有效性、安全性、舒适性等方面进行评估。(随访评估内容可参考附录Ⅲ)
四、统计分析报告和临床试验报告
(一)统计分析报告
应将所有中心的数据合并在一起进行统计分析,并出具总的统计分析报告。应对随机对照部分进行统计分析,并出具相应的统计分析报告。统计分析报告应至少包括如下4部分内容:
1.临床试验完成情况描述:包括临床试验概况(筛选人数、入组人数、完成试验人数、失访/退出/剔除人数等)。
2.基线描述:应对所有入选受试者的基线人口统计学指标、生命体征及其他相关病史指标等进行描述。
3.疗效/效果评价:应对全分析集和符合方案集分别进行统计分析。
4.安全性评价时,应对接受过治疗的所有受试者进行分析,不能遗漏所有发生的任何不良事件。同时,详细描述每一病例出现的全部不良事件的具体表现、程度、预后及其与研究产品的关系。
(二)临床试验报告
各临床试验单位在遵循统一临床试验方案基础上,由临床试验申办者与研究者根据基于所有入选受试者的总的统计分析报告,出具临床试验报告。各临床试验单位出具临床试验小结。各临床试验单位不需要单独出具分中心统计报告。临床试验报告内容包括:试验目的、试验假设、主要评价指标、评价方法、对照品、入选/排除标准、样本量及计算依据、受试者资料、试验质量控制措施、数据管理及质控措施、试验结果、伴随治疗、不良事件、并发症及其处理、试验结论、适用范围、禁忌症和注意事项、存在问题及改进意见等。
此外,需注意以下问题:
1.临床试验报告应与临床试验方案保持一致。
2.明确所有病例是否全部完成随访,所有接受了器械治疗的病例是否均纳入最终的统计分析,失访病例需明确失访原因。
3.报告所有不良事件发生的时间、原因、后果及与试验用器械的关系,对于所采取的处理措施需予以明确。
五、参考文献:
1.《医疗器械注册管理办法》(国家食品药品监督管理总局令第4号)
2.《医疗器械临床试验质量管理规范》(国家食品药品监督管理总局 中华人民共和国国家卫生和计生委员会令第25号)
3.《关于发布医疗器械临床评价技术指导原则的通告》(国家食品药品监督管理总局通告2015年第14号)
4.瞿佳。《视光学理论和方法》人民卫生出版社 2004
5.葛坚。《眼科学》人民卫生出版社 2005
6.Premarket Notification (510(k)) Guidance Document For Daily Wear Contact Lenses. (美国FDA)
六、起草单位
本指导原则由国家食品药品监督管理总局医疗器械技术审评中心起草并负责解释。
来源:国家药监局

为你推荐
 资讯
资讯 BMS 2.86亿美元收购了一家CAR-T疗法公司
近日,百时美施贵宝(BMS)宣布将以每股5 00美元的全现金交易方式收购2seventy bio(TSVT US),总股本价值约为2 86亿美元,交易预计将在2025年第二季度完成。
2025-03-13 16:28
 资讯
资讯 启明医疗正式复牌:以长期主义开启高质量发展新阶段
此次复牌标志着启明医疗彻底解决了公司治理问题,并重新建立了内部控制体系,是公司回应市场关切、重塑行业信心的关键一步。
2025-03-13 09:13
 资讯
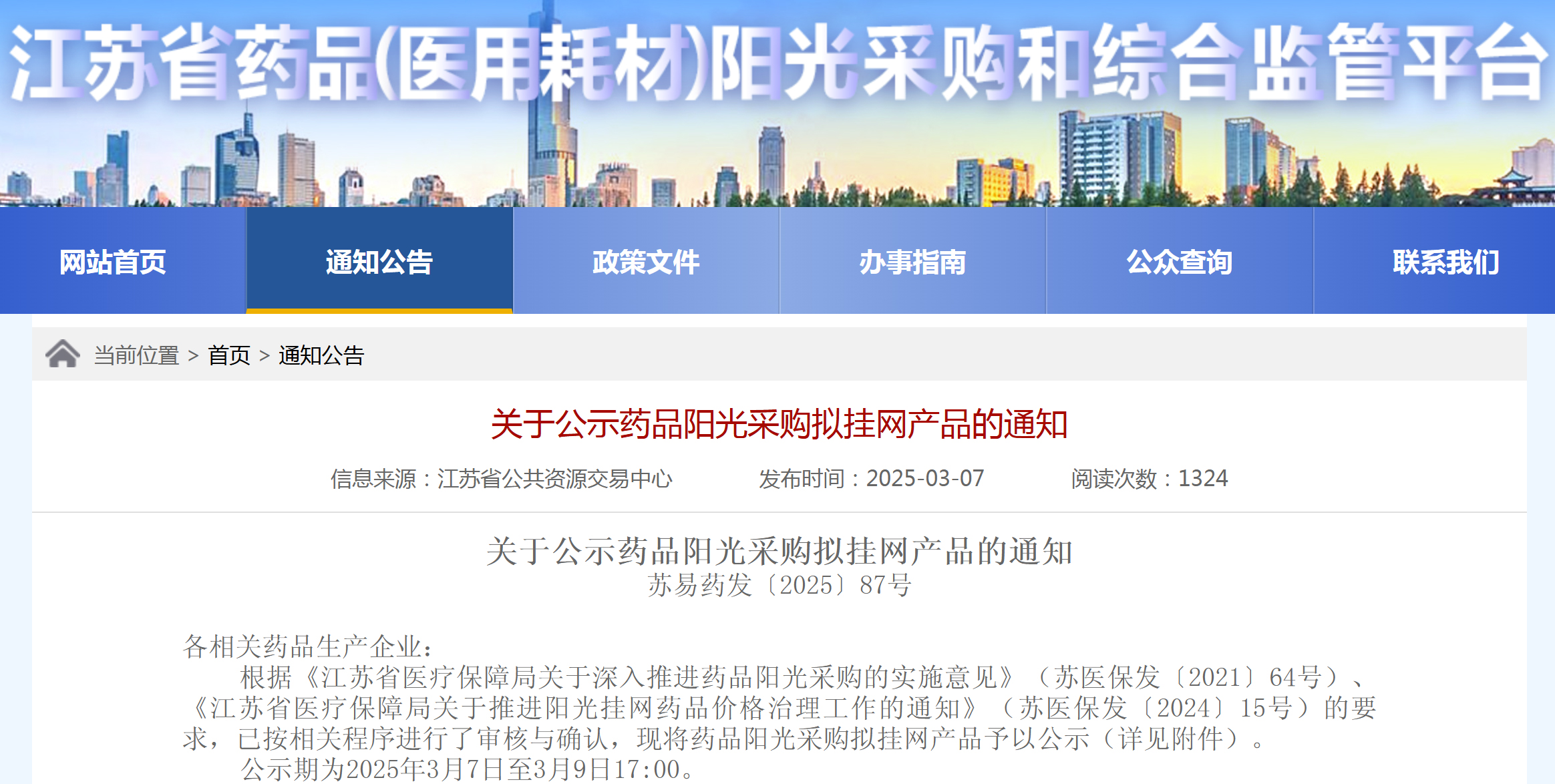
资讯 原生多模态+千亿级数据训练,蚂蚁医疗大模型拿下MedBench测评“双料”冠军
近日,记者发现,国内权威医疗大模型评测平台MedBench在官网更新了榜单。多个医疗AI产品及研究团队入榜,其中蚂蚁AI健康管家团队研发的蚂蚁医疗大模型以评测榜单97 5、自测榜单...
2025-03-12 15:47

 资讯
资讯 业绩增速持续承压,爱美客超13亿元收购了一家韩国医美公司
近日,爱美客发布公告称,基于公司战略规划和经营发展需要,其全资子公司 Imeik(HK)Limited(简称“爱美客香港”)与 Aisheng Shourui (HK) Limited(简称“首瑞香港”...
2025-03-12 10:47

 资讯
资讯 重见光明,聂爷爷的笑容回来了——重庆爱尔助孤寡白内障患者重燃生活希望
对于陈茂盛院长和重庆爱尔眼科医院(总院)的医护人员来说,聂爷爷的重见光明正是他们不断追求的动力。
2025-03-12 09:41
 资讯
资讯 无双医疗完成C轮近1.5亿元融资,加速心脏节律管理创新产品研发和商业化
本轮融资由天士力资本领投,现有股东启明创投、苏高新金控、康裕资本继续加持,为无双医疗在心脏节律管理(CRM)领域的创新产品研发和商业化进程注入了强劲动力。
2025-03-12 09:38
 资讯
资讯 卫美健康完成A轮亿级融资,加速县域基层医疗大模型应用落地
本轮融资资金将主要用于卫美健康“奇点医问”医疗大模型的研发投入与迭代进化,加强医疗细分领域的算法研究,构建升级全国服务网络体系,进一步扩大卫美健康在县域以及基层AI医...
2025-03-12 09:19
 资讯
资讯 突发,上市公司双成药业宣布终止跨界重组
昨日晚间(3月10日),双成药业发布公告称,公司原拟以发行股份及支付现金的方式向奥拉投资、Win Aiming等25名交易对方购买其持有的宁波奥拉半导体股份有限公司100%股份,并拟...
2025-03-11 13:44

 资讯
资讯 百时美施贵宝公布颂狄多®(氘可来昔替尼)POETYK PsA-2 III期试验最新数据 证实其在治疗成人银屑病关节炎中优于安慰剂
治疗第 16 周时,颂狄多治疗组患者的 ACR和 PASI应答率显著高于安慰剂组,且患者报告生活质量有更明显改善。与安慰剂和阿普米司特相比,颂狄多耐受性良好,安全性特征与既往...
2025-03-11 09:38
 资讯
资讯 华东医药全球首个卵巢癌ADC爱拉赫®补充申请获受理
申报适应症为用于既往接受过1-3线系统性治疗的叶酸受体α(FRα)阳性的铂类耐药的上皮性卵巢癌、输卵管癌或原发性腹膜癌成年患者。
2025-03-10 19:36

 资讯
资讯 用于治疗复发或难治性多发性骨髓瘤,辉瑞靶向免疫疗法易瑞欧(埃纳妥单抗)在华获批
今日(3月10日),辉瑞公司宣布,靶向免疫疗法易瑞欧®(埃纳妥单抗)获国家药品监督管理局附条件批准,适用于既往接受过至少三线治疗(包括一种蛋白酶体抑制剂、一种免疫调节剂...
2025-03-10 12:07
 资讯
资讯 一款国产肺癌创新药头对头击败奥希替尼
日前,同源康医药发布公告称,其自主研发的第三代EGFR抑制剂TY-9591(商品名:卡达沙)在对比奥希替尼(商品名:泰瑞沙)作为一线治疗EGFR突变肺癌脑转移的关键II期临床试验中,...
2025-03-10 10:55
 资讯
资讯 “AI+创新药”第一股云顶新耀开拓mRNA肿瘤治疗性疫苗新蓝海,在国内推进至临床阶段
近年来,AI 赋能创新药研发已成为全球生物医药行业的重要趋势,尤其在 mRNA 疫苗领域,AI 更是成为提升研发效率与精准度的核心驱动力。港股创新药企云顶新耀(HKEX 01952 ...
2025-03-10 09:29

 资讯
资讯 “全力治愈的春天音乐会”乳腺癌公益项目在南京暖心启航
3月7日,南京国民小剧场内,一场特殊的“疗愈音乐会”正在温暖上演。没有冰冷的医学术语,没有沉重的疾病阴霾,取而代之的是歌声、琴声、孩童的欢笑和患者含泪的拥抱。
2025-03-08 18:03