
测序技术已大大超出了Carlos Bustamante、Stephen Kingsmore和John Mattick三位博士的预期。如果你在他们职业生涯刚开始时询问他们,是否有一天我们能在一天内测序人类全基因组,他们的反应分别是:“疯言疯语!”,绝对不可能“以及”做梦也不敢想“。
尽管测序创新的速度让他们惊讶,但每个人都迅速采用了新一代测序(NGS)和如今的群体测序,以便推进他们的科研和转化工作。作为遗传学和生物医学数据科学的教授和斯坦福计算机、进化和人类基因组学中心的创始主任,Bustamante博士正利用群体测序来了解古代和种族亚群中的遗传变异。Kingsmore博士最近新任Rady儿童医院基因组医疗研究所的总裁兼CEO,他正利用测序来开发儿童基因组医疗的证据基础。作为Garvan医学研究所的执行主任,Mattick博士正带头利用群体测序数据开展研究和临床应用。
iCommunity此次与Bustamante、Kingsmore和Mattick三位博士对话,聊聊他们的团队如何利用高通量的人类全基因组和群体测序来推进科研和转化研究,融合”组学“和表型数据的数据库的要求,以及将这一信息转化成对临床环境有用的格式所面临的挑战。
在您刚成为科学家时,测序技术是什么样的?
John Mattick (JM):我对测序的第一印象是看见放射自显影图上的条带。这是分子生物学的早期。我们正在克隆和测序基因。我当时认为,我们是高手。我们只能从胶上读取几百个碱基,之后条带挨得太近无法区分。我们组装成1-2 kb长的序列,每条序列都能发一篇论文。现在回头看,这似乎太原始了。
Stephen Kingsmore (SK):我的测序体验是从放射性的p32标记以及琼脂糖和聚丙烯酰胺凝胶开始的。一个了不起的测序反应是150个核苷酸,而那要花去大半天。
Carlos Bustamante (CB):我成为科学家时,自动化测序正在开发中,因此我开展了一些手动测序,之后在第一代测序仪上进行大量的测序。我初次体验是在史密森学会实习时,他们刚刚建立了分子系统学实验室。那时,测序多名个体的几个基因可是大工程。
当工具改进时,您的测序方法如何改变?
CB:一开始,我们将每个片段的数据都看得很宝贵。当Celera开始进行早期的外显子组测序时,他们对20万个样品进行PCR,并测序39个人的2万个基因。我想,”这是一个数据集!我们一直在等待这个。“我们停下了手头的工作,花了4-5年的时间来研究这39个外显子组,并发表了8-9篇论文,以不同的方式分析数据。这种思维模式已经被颠覆了。如今,我们利用NGS不断地快速生成数据,然后担心它意味着什么。
当新一代测序(NGS)工具被引入时,您多快将其引入研究?
CB:NGS快速成为我们研究中的重要工具。我们是猕猴和猩猩基因组计划中的一部分,其中我们分析多态性数据。我们也是千人基因组计划最初的分析小组之一,设计美洲的采样,确定2x-4x测序的价值,以及变异频率的界限。
SK:NGS系统上市没多久,我们就开始使用了。那是激动人心的日子。我们将邮件收发室改为NGS实验室。关于人类基因组,人们知道得还不多,因此我们每项研究都在发现新东西。
JM:多年来,我一直是基因组学新技术的早期采用者。与Craig Venter一样,我是Molecular Dynamics的Megabase测序仪的早期客户之一。Garvan研究所是最早购买HiSeq X? Ten系统的3家机构之一。
”获得精确变异信息的唯一的方式是获取数十万个基因组的准确变异信息,这样我们才能评估我们所看到的每个变异的频率。“
您早期的测序工作如何影响您目前研究的重点?
CB:在早期,我们研究感兴趣的基因中的多态性和变异。在我的博士论文中,我分析了当时最大的基因组数据集,它包括对多只果蝇测序的25个果蝇基因和对多株植物测序的15个拟南芥基因。我们查看氨基酸的差异以及有利和有害突变的积累。从那时起,我开始考虑创建人类序列的大型数据集,这样我们就能以同样的方式分析。
SK:在国家基因组资源中心的时候,我们利用早期NGS来测序植物和病原体的转录组,后来是基因组,并开始测序人类样品。我们中的一些人认识到,我们在科研环境中开展的研究不久将会影响医疗保健。在环顾全国之后,3个人去了堪萨斯城的儿童慈善医院,建立第一批儿科基因组学医疗中心,并开始进行转化研究。我目前在Rady儿童医院的基因组科学研究所,在这里我们将更进一步,关注基因组系统医疗在加利福尼亚州最大儿童医院中的大规模实施。
JM:高通量测序对认识人类基因组的转录复杂性有巨大影响。NGS加快了我们深入转录组的能力,让我们能够探索非编码转录本的奇妙世界,它在发育过程中以精确的方式从不同的细胞和组织的基因组中涌出。我认为人类基因组就像特别的。ZIP压缩文件。人类基因组的转录复杂性至少比基因组本身高了一个数量级,它能够以不同的方式解压缩,在不同的时间,在不同的细胞中呈现出不同的编码和非编码RNA的表达和剪接模式。没有高通量测序,我们将无法探索这个世界。
”在基因组学的新世界,每个学生、每个博士后、每个实验室和每个部门都需要有能力去处理大数据。“
您现在如何使用NGS?
CB:NGS已经为群体基因组学开辟了新道路。我记得曾在冷泉港会议上,我意识到千人基因组计划应包含混杂的基因组。人们质疑这一点,但我认为,若要分析和开展跨种族和多种族的研究,我们需要弄清楚如何理解混杂基因组。
我们参与临床基因组资源(ClinGen)联盟的一个原因是汇集临床基因检测数据,并削弱意义不明的变异(VUS)的比例,这在某些少数人种群体中更高,仅仅是因为没有太多序列可供分析。NGS让继续追踪这些GWAS hit变得廉价且轻松。我们发现的每个氨基酸改变都是确凿的证据。显然,如果我们真的想要开发让每个人都受益的基因组医疗,我们需要拓宽人类DNA研究中的种族代表。
SK:我们的重点在全基因组测序(WGS),因为这是终极的分子检测。WGS如今更快了,我们与Illumina合作,开发出一种方法,让我们能够在26小时内解码和分析整个人类基因组1。我们的目标是到明年年中,向我们新生儿和儿科重症监护室(NICU和PICU)中每个无法确诊的儿童提供快速NGS,并开展临床研究,以确定基因组医疗在儿科住院和门诊环境中的临床效用和成本效益。
HiSeq X系统让您实现了哪些研究?
CB:群体测序是我一直希望达到的顶峰 – 分析多个人类基因组。我们在开展大规模的群体测序研究,以它们作为基线来回答重要的群体遗传问题,并分析结果,为临床医学带来新方法。例如,我们综合利用大规模的基因分型和测序,在秘鲁开展一项子痫前期的研究,并研究一下高原适应,因为它与子痫前期相关联。
SK:利用HiSeq X系统,基因组变得便宜很多,因此我们能够测序更多的家系。目前有8000种已命名的遗传病,我们及其他人都强烈感觉到,NGS将改变我们鉴定遗传病的能力。我们希望利用HiSeq X和Illumina SeqLab设施,逐步开发证据基础来支持这一点。
”我们最大的挑战就在于如何分享群体测序数据。“
JM:Garvan研究所是最早将基因组学推向研究工作的中心的研究所之一,而不是作为传统分子生物学的延伸。随着基因组测序的巨大进步以及随之而来的成本下降,开展群体测序并将基因组学推向科研和临床的中心已经在经济上变得可行。
HiSeq X系统如何让转化和科研工作融合,这是非同寻常的。我们一直与全世界的研究人员合作。HiSeq X Ten系统的表现很出色。
除了研究单基因疾病,我们也在大型研究项目中使用群体测序,包括癌症、糖尿病、骨质疏松症、免疫学疾病、神经退行性和神经精神疾病,以及衰老。作为国际癌症基因组联盟(ICGC)的一部分,我们正开展癌症分层研究,并利用NGS阐释癌症基因组,评估家族癌症风险的遗传元素。我们对1型糖尿病的患者进行测序,以发现一生中状况良好的患者与后期患有严重并发症(如肾衰竭)的患者之间的遗传差异。在我们的衰老研究中,我们正利用群体测序来研究数千名个体,他们年事已高,但没有心血管疾病、癌症、认知能力衰退或神经退行性疾病的任何迹象。我们正在开发风险去除队列,它们可作为对照,用于罹患此病的患者的研究。利用HiSeq X Ten测序能力的其他项目包括研究患有心脏、线粒体和阿尔茨海默病的群体。
您在分享群体测序数据时有何挑战?
CB:我们最大的挑战就在于如何分享群体测序数据。NIH及其他机构如今命令研究人员分享他们的数据。不幸的是,这对临床数据而言是不正确的。大多数医院都没有真正的数据分享原则。我们也生活在一个互相连通的世界,这让患者对分享信息感到不自在。因此,国际基因组学健康联盟及其他机构开发前瞻性知情同意、隐私程序以及数据管理和透明度上的最佳实践将是很有价值的。
SK:当我们在Rady儿童医院测序基因组之前,父母必须签署知情同意书。知情过程的一部分是同意我们能够发表基因组。我们去除识别信息,这样就没有信息能够将基因组与儿童或父母相关联,然后信息就可以从美国国家生物技术信息中心(NCBI)的基因型和表型数据库(dbGaP)中获取,这是一个私人的数据库。研究人员只有在向NIH申请,并很好地解释他们为什么需要获取这个信息之后,才能得到数据。这似乎在隐私方面的担心和其他研究人员能够研究公开基因组的好处之间达到了良好的平衡。
不幸的是,并非所有医院都有一个适当的基因组共享知情同意过程。临床研究人员需要人类全基因组序列信息来确定基准。他们想看看变异在基因组中有多常见。唯一的方式是获取数十万个基因组的准确变异信息,这样我们才能评估我们所看到的每个变异的频率。
”随着基因组测序的巨大进步以及随之而来的成本下降,开展群体测序并将基因组学推向科研和临床的中心已经在经济上变得可行。“
将WGS、表观基因组、转录组及其他基因组和表型数据相整合,获得不同的基因组快照,有何价值?
CB:开展各种类型的组学分析,RNA-Seq、甲基化组测序等,具有重要的价值。我们仍不太理解人体的调控网络。我们如今在开展和整合组学数据吗?我想,这进行得很慢,部分原因是测序要比解释简单得多。
SK:泛组学无疑具有价值,其中我们正获取全基因组数据,并将它与深度表型组、表观遗传、基因表达、代谢组和蛋白质组的数据结合在一起。测序基因组并不是游戏的结束,而是一个伟大的开始。我们开始了解,我们需要什么才能带来精准医疗。例如,我们不知道我们在基因组中发现的大多数变异在功能上意味着什么。因此,我们不能自信评估,它们是否让人体产生改变。显然,我们需要更多类型的数据,能够大规模开展这种评估。
JM:临床研究和医疗的未来将围绕着大数据的整合。这不仅仅是个别的及合并的基因组数据集。这些将逐渐与转录组、表观基因组、蛋白质组,以及最重要的表型数据相融合,创建高度关联、富含信息的数据集。医疗正在快速向大数据迈进,而数万个、数十万个基因组序列的获得将使其加速。它即将改变一切。
生物信息学和数据库对挖掘群体测序的全部价值有多重要?
CB:从一开始,我们就很清楚需要将测序与分析工具相结合,才能理解所有数据。通过关联和分析表型及基因型信息,我们开始揭开在静态数据中看不到的模式。人们有一种乐观的态度,如果我们能够以更严格的方式测定表型和暴露,我们就能够收集到海量的数据,帮助我们发现遗传关联。
JM:我认为,生物信息学框架和数据库对整个工作很关键。它将基因组数据与正交数据集相整合,以提取宝贵的信息。我们确定的遗传模式将有助于了解个体在临床中的情况,并通过元数据的分析,了解就疾病模式、并存疾病而言的整个健康体系。
群体测序并不是一件轻松的事情。在过去1-2年,我们投资了1000万美元来建立计算管道。整个组装管道有一个不断扩大的60个人团队,开展测序、组装数据、检出变异和群体之间的差异,并将数据与表型数据相关联。
在基因组学的新世界,每个学生、每个博士后、每个实验室和每个部门都需要有能力去处理大数据。这终将不是专家的事。它对整个研究和医疗工作都很关键。这是一个数据驱动的世界,我们正冲向它。
SK:我们在最近的研究中认识到生物信息学的价值,这项研究比较了WGS和传统基因检测在确定危重新生儿的孟德尔疾病中的效果2。为了分析数据,我们开发了一些新的生物信息学工具。论文证明了基因组测序的实用性,但我们需要基因组学的临床价值的进一步证据。我们还需要一种简化的方法将结果告知医生,不仅关系到诊断,还关系到NGS数据如何提供治疗决策。
”泛组学无疑具有价值,其中我们正获取全基因组数据,并将它与深度表型组、表观遗传、基因表达、代谢组和蛋白质组的数据结合在一起。“
您需要哪种类型的数据库?
JM:我们需要全国水平的基因型/表型关联数据库,它们由卫生部门维护,可供认可的研究人员和医生查询。它们必须是全国的数据库,因为每个管辖区存在特有的法律及其他要求。它们需要以某种方式与一个全球数据库相关联,这样一个国家产生的数据可在其他地方使用,并以多维度的方式探索,以便推进我们对人类生物学和疾病的了解。
”我认为最广泛意义的群体规模测序将从儿童开始,可能是在出生时,以取代现在的Guthrie检查。“
创建这些数据库需要多长时间?
JM:我们不可能在一夜之间对全世界的每个人测序,但我相信,十年内我们将有大型的基因组数据库。基因组数据将逐渐成为病历的一个标准部分。在理想情况下,我们将在云端拥有充分审核、基于证据的基因型/表型关联数据库,它们将被维护,并不断更新全国资源。
最初的应用将是对有着严重遗传缺陷的个体进行测序,因为我们能够快速从半数病例中诊断出致病突变。癌症分层将是一个重要领域,让医生能够确定疾病的分子基础,从而更有效地治疗疾病。第三个领域将是检测药物副作用的遗传标志物,因为这对每个国家的医院体系是个巨大的负担。我们能够通过基因组信息预测和避免大部分的副作用。
我们正建议澳大利亚医疗系统对每个带有发育和/或智力障碍的人进行测序,作为一线治疗。我预计,这将在2-5年内变得常规。我认为最广泛意义的群体规模测序将从儿童开始,可能是在出生时,以取代现在的Guthrie检查。新一代的儿童将是基因组一代,在他们身上选择性地开展基因组测序和分析,之后随着技术和信息的价值改善,再逐步广泛应用。
您认为WGS将成为一种常规的临床检测吗?
JM:测序距离常规应用(即体检的一部分)并不遥远。测序费用将不断下降,这使得人们能够再次分析以提高某个人的基因组原始数据的准确性,融入表观基因组和转录组数据,或查看体细胞变异。随着我们更深入了解基因组中的变异对生物学和医学意味着什么,测序的价值将不断提高。测序在医学中的更广泛使用如今受到数据库的丰富性和质量的限制,这些落后于信息的分析。
值得一提的是,美国医学遗传学家学会(ACMG)强制报告56个基因,因为这些可能与患者未来的健康有重要的关系。我们将开始查看充分验证的基因集合,无论是强制报告的,还是这一领域的机构自信报告给医生和患者的,随着时间的推移,这个列表将不断扩展。
SK:我们有着新生儿筛查项目的丰富传统,其中每个婴儿在出生时都会进行足跟采血,检测29种疾病。一些美国的研究小组开始研究,如果我们将足跟采血换成基因组测序,那将会提供其他哪些信息。我们还不知道。
”群体测序将让我们发现和鉴定与不良反应有关的临床可行动变异的整体等位基因频率。“
人类全基因组数据是否让我们离个性化医疗更近?
CB:我认为,基因组测序最终将成为常规医疗的一部分,以及人们电子病历中的一部分。这是一个有趣的时代,因为我们有点处在过渡阶段。测序技术已经成熟,而人们在开展高通量测序,很快将常规开展群体测序。
我们需要提出一个协商好的计划,来集合这些数据,分析它们,并尽快将其转化成健康收益。最终,我们需要为公众提供良好的投资回报。
SK:未来,测序结果将带来治疗的改变。一般来说,诊断领域是属于病理学家和检验师的,而医疗实施才是医生的责任。对于基因组医疗,这两项将融合。这将是一个挑战,因为没有一方习惯让另一份来参与这些任务或信息。
JM:我认为问题在于我们对基因组的了解还很有限。如今,我们只能准确报告蛋白编码序列中的一些变异的影响。从文献中收集足够的证据和数据,以便自信检出基因组其他部分中可能有医学意义的突变或变异,这是一个大工程。通过群体测序创建的全球大型数据库将支持这一工作。这些数据库将包含反映一系列突变和表型特征的序列,并允许人们通过查询来确定新样品是否反映了数据库中已存在的症状和突变。
群体测序的数据将如何改变医疗?
JM:群体测序将对医疗产生深远的影响,将其从危机管理的艺术转变为良好健康的科学。我们知道,个体的基因组变异和我们的遗传特质影响了我们现在的健康,并带来了未来疾病的风险,无论是2型糖尿病、癌症、类风湿性关节炎,还是阿尔茨海默病。对许多病例而言,有备无患,这让医生和患者能够实施策略,以降低、避免或准备这些可能性。
SK:我研究儿童的罕见遗传病,它们只是遗传的。我们如今有能力做出快速诊断,因此,这些疾病可能在开发和制造新药上具有成本效益。我们的希望是,基因组学在诊断复杂疾病上将逐渐变得与单基因疾病同样宝贵。这也许需要几十年才能赶上,但群体研究对缩小这一差距将是非常重要的。关于群体研究,激动人心的一件事是我们开始根据遗传学来重新定义我们描述疾病的方式,而不是根据症状。
JM:群体研究将为治疗药物的开发提供信息,特别是在鉴定不良反应的遗传学上。美国一年有10万人死于处方药的不良反应3。在澳大利亚,至少2-3%的入院是由于处方药的不良反应4。
CB:例如,Abacavir是一种重要的HIV药物,而研究人员已鉴定出一种与Abacavir过敏相关的HLA变异。这个变异在非洲和欧洲人中的流行程度很低,但在印度和亚洲的某些群体中却达到20%的频率5。如果携带变异的患者服用一次Abacavir,他们会变得非常虚弱。如果他们服用两次,则会死亡。群体测序将让我们发现和鉴定与不良反应有关的临床可行动变异的整体等位基因频率。瓶颈在于让医生理解药物代谢信息,这样他们将知道选择药物A还是药物B,或者将剂量减半或加倍。
JM:制药公司也开始利用群体测序来鉴定过去药物试验中的异常响应者。如果他们能够将群体分层,并确定响应者的特定遗传背景,则他们能够分析相关的生物化学通路。他们不仅在挽救失败的药物,也在挽救响应患者,带来有效且可能救命的治疗。
”特别是在美国,我们需要对那些健康结果最差的种族群体进行群体测序,这样保健上的负缺口才不会扩大。“
种族亚群的测序有多重要?
CB:我们掌握技术来开展群体测序,这真是棒极了。然而,我们需要协调工作,让研究继续在种族亚群上开展。没有的话,重点将仍是测序大量的同质群体,如芬兰人或冰岛人。尽管这些工作很重要,但它们的收益将无法转化到所有群体。特别是在美国,我们需要对那些健康结果最差的种族群体进行群体测序,这样保健上的负缺口才不会扩大。这带来了一个挑战,因为不会有高级的行动来资助这些工作。美国政府的精准医疗行动是一项伟大的工作,但它不能与英国及其他国家的工作相比。特别是中国,它认为基因组学是他们发展计划中主要的一块。
”最终,基因组信息将自动从智能设备报告到云端。这将带我们进入一个从未梦想过的境界。“
1,000美元基因组已经或将要产生哪些影响?
SK:好消息是,1000美元基因组存在于群体测序中。在临床保健中,我们需要的是快速基因组测序的成本也降低至1000美元基因组的水平,这还没有实现。
JM:1000美元基因组是一个实际且心理的临界点。它改变了我们考虑技术的方式以及我们认为哪些是可能的。它激发了临床和科研活动的整合,以一种我们从未想过的方式。人们如今认识到,基因组学正从一个研究工具逐渐转变成一个日常的临床分析工具。
当您刚成为科学家时,您是否相信有一天人类全基因组测序将在一天内开展?
CB:我可能会说,那是不可能的。疯言疯语!
SK:绝对不可能。即使让我回到我使用第一台Solexa系统测序时,我也不敢期望,我们能够如此快速地大量产生基因组。
JM:做梦也不敢想。在20世纪下半叶,我们才刚刚懂得DNA长什么样,基因长什么样,并开发出原始的基因组分析工具。那时,我们所做的一切都被认为是处在前沿,的确是这样。如今,我们以超乎寻常的速度前进。21世纪将是生物学和医学的世纪。NGS与大数据的整合仍将展开,并带来可预见的未来。最终,基因组信息将自动从智能设备报告到云端。这将带我们进入一个从未梦想过的境界。这是一个精彩绝伦且激动人心的时代。我们很感激Illumina这样的公司,从技术上引领这一切。
参考文献
1. Miller NA, Farrow EG, Gibson M, et al. A 26-hour system of highly sensitive whole genome sequencing for emergency management of genetic diseases. Genome Medicine. 2015; 7(1) 100. do: 10.1186/s13073-015-0221-8.
2. Willig LK, Petrikin JE, Saunders CJ, et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir Med. 2015; 3(5):377–387.
3. Preventable Adverse Drug Reactions: A Focus on Drug Interactions. U.S. Food And Drug Administration. www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm110632.htm. Accessed May 16, 2016.
4. Roughead L, Semple S, Rosenfield E. Literature Review: Medication Safety in Australia. www.safetyandquality.gov.au/wp-content/uploads/2014/02/Literature-Review-Medication-Safety-in-Australia-2013.pdf. Published August 2013. Accessed May 16, 2016.
5. Puthanakit T, Bunupuradah T, Kosalaraksa P, et al. Prevalence of human leukocyte antigen B*5701 among HIV-infected children in Thailand and Cambodia: implications for abacavir use. Pediatri Infect Dis J. 2013; 32(3): 252–253.
来源:Illumina

为你推荐
 资讯
资讯 带状疱疹疫苗“遇冷”,百克生物2024年净利润腰斩
近日,国内疫苗龙头企业百克生物发布2024年年报,数据显示,其报告期内实现营收12 29亿元,同比下降32 64%;归属于上市公司股东的净利润2 32亿元,同比下降53 67%。对于营收...
2025-04-23 12:59





 资讯
资讯 重庆常用药联盟接续集采中选结果
近日,重庆常用药联盟接续集采中选情况公布,该联盟由重庆牵头,联合湖北、广西、海南、贵州、云南、青海、宁夏、新疆及新疆生产建设兵团等十省(区、市)开展的药品集中带量采...
2025-04-21 18:48

 资讯
资讯 全周期智控慢病,诺和诺德与京东健康开启战略合作
2025年4月21日,全球领先的生物制药公司诺和诺德与京东健康在北京正式签署战略合作协议,标志着双方在糖尿病和体重管理领域的合作进入新阶段。依托诺和诺德百年深耕慢病领域的专...
2025-04-21 15:57
 资讯
资讯 康方生物1类新药依若奇单抗上市申请获批,用于中重度斑块状银屑病成人患者
该药是我国第一个且唯一获批上市的IL-12 IL-23“双靶向”单克隆抗体新药,是康方生物自身免疫性疾病领域首个获批上市的一类新药。
2025-04-21 13:39
 资讯
资讯 阿斯利康乳腺癌1类创新药卡匹色替片中国获批
该药适用于联合氟维司群用于转移性阶段至少接受过一种内分泌治疗后疾病进展,或在辅助治疗期间或完成辅助治疗后12个月内复发的激素受体(HR)阳性、人表皮生长因子受体2(HER2)...
2025-04-21 11:02
 资讯
资讯 辉瑞宣布终止一款口服GLP-1减肥药的临床开发
近日,辉瑞在其官网宣布,决定终止开发口服胰高血糖素样肽-1受体(GLP-1R)激动剂Danuglipron(PF-06882961),原因系在一项有关用药剂量的临床试验中,一名患者出现了可能由该...
2025-04-21 10:29
 资讯
资讯 福建省医保局印发单列门诊统筹支付医保药品目录(2024年版)
根据2024年6月发布的《福建省医保药品单列门诊统筹支付管理办法(试行)》,为了让参保患者无需住院、在门诊就医也能用上国家谈判药品、享受医保待遇,将适用于门诊治疗、使用周...
2025-04-20 13:34
 资讯
资讯 首批中国消费名品名单,医药健康企业有哪些?
近日,工业和信息化部办公厅发布首批中国消费名品名单,分为中国消费名品名单和中国消费名品成长企业名单。首批中国消费名品名单共包括93个企业品牌和43个区域品牌。中国消费名...
2025-04-20 11:17
 资讯
资讯 携手共绘“个性化近视手术”新蓝图:爱尔眼科与爱尔康启动100家医院全光塑技术战略合作
双方将以技术共享为核心,以人才培养为支撑,以科研协作为纽带,全力推进屈光手术标准化诊疗体系建设,加速前沿技术在临床领域的普及应用
文/ 屈慧莹 2025-04-19 23:35
 资讯
资讯 CDE:简化港澳已上市传统口服中成药内地上市注册审批申报资料及技术要求
允许香港、澳门特区本地登记的生产企业持有,并经香港、澳门特区药品监督管理部门批准上市且在香港、澳门特区使用15年以上,生产过程符合药品生产质量管理规范(GMP)要求的传统...
2025-04-18 18:54
 资讯
资讯 君德医药完成近亿元A轮融资,加速推进创新药械组合平台建设与产品上市
本轮融资主要用于首个减重口服器械的注册及生产销售,以及加速多个核心创新药械组合技术平台的产品管线研发进程。
2025-04-18 14:34
 资讯
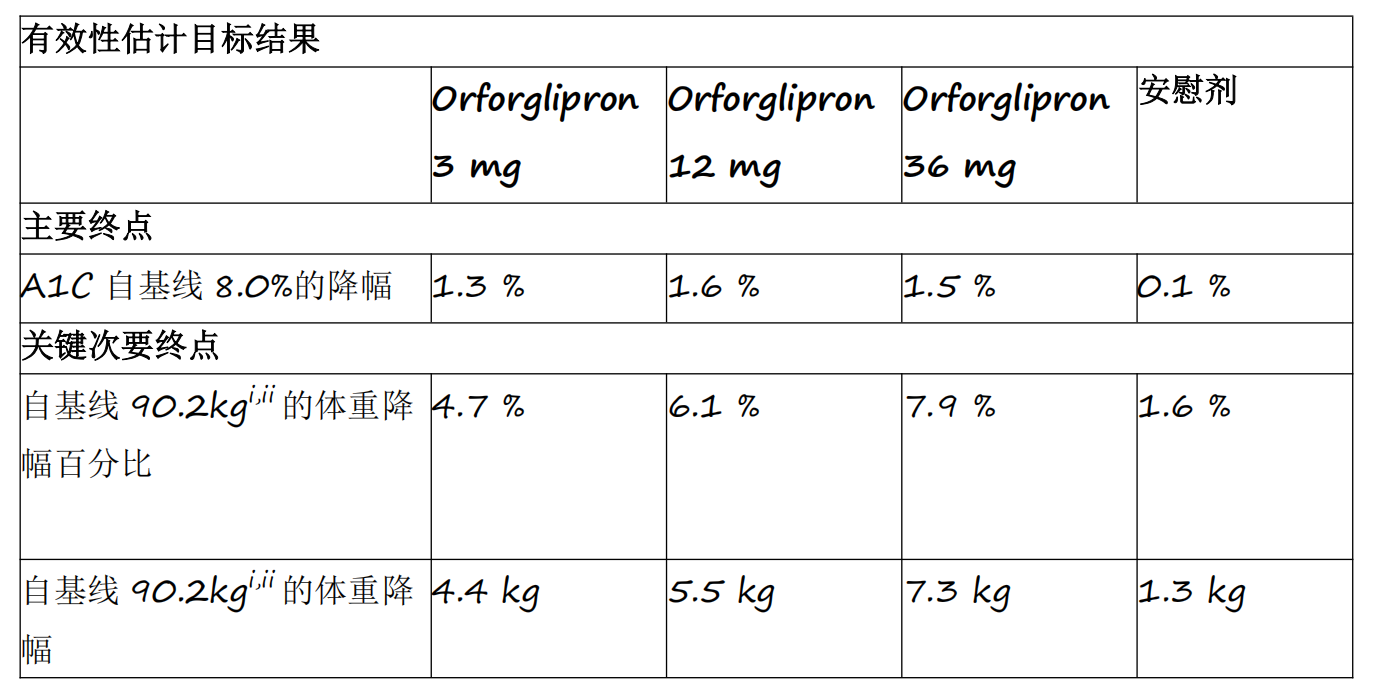
资讯 礼来首个小分子口服GLP-1RA药物orforglipron 3期临床研究成功
Orforglipron是首个成功完成3期临床研究的小分子GLP-1类药物,各剂量组平均A1C降幅为1 3%至1 6%
2025-04-18 14:12
